

MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
New York University Medical Center
New York, New York
1980
Preface
There is a large and rapidly growing body of literature on the importance of magnesium in biochemical and physiological processes. There is also much evidence that magnesium deficiency, alone and in combination with agents that interfere with its utilization, is associated with functional and structural abnormalities of membranes, cells, organs, and systems. The manifestations of the changes caused by magnesium deficiency depend upon its extent and duration and on variable factors. Among the conditions that increase the risk of magnesium deficiency are (1) metabolic factors that affect the absorption, distribution, and excretion of this mineral; (2) disease and therapy; (3) physiologic states that increase requirements for nutrients; and (4) nutritional imbalances. Excesses of nutrients that interfere with the absorption or increase the excretion of magnesium-such as fat, phosphate, sugar, and vitamin D-can contribute to long-lasting relative magnesium deficiency. All have been implicated in several of the diseases considered in this book. Whether their influence on the need for magnesium is a common denominator remains to be investigated further.
Unfortunately, means of diagnosing clinical magnesium deficiency of a lesser degree than that associated with overt signs such as convulsions or cardiac arrhythmias or other electrocardiographic changes are not readily accessible. Plasma magnesium levels are unreliable as an index of its cellular inadequacy. More complicated means of evaluating the magnesium status are considered in the Appendix, as are their limitations and need for convenient determinants. Until magnesium clinical methodology is improved and made available, the importance of correcting magnesium deficiency in man's diet and of preventing intensification of a deficit when needs are increased by physiologic or pathologic processes and drugs will have to be inferential-based on experimental and epidemiologic observations. Because magnesium has pharmacologic activities that have been recognized for many years, demonstration of the correction of abnormal acute neurologic and cardiac signs (even though such signs are characteristic of acute magnesium deficiency) are not readily accepted as evidence that magnesium deficiency can contribute to diseases in which such magnesium-responsive signs are seen. With notable exceptions, there has been clinical neglect of magnesium in most medical centers and certainly in private practice. This is unfortunate because many of the pathologic changes produced by experimental magnesium deficiency or loss resemble many of those of chronic diseases that are responsible for intractable medical problems.
This book develops the premise that magnesium deficiency during gestation is more common than generally believed and that it may be contributory to some disorders of pregnancy and infancy. It draws parallels between cardiovascular and skeletorenal lesions of infancy and childhood and those produced by magnesium deficiency-especially when intensified by dietary excesses of vitamin D and of phosphate, which are commonly consumed in the United States and other Occidental countries. It suggests that the most severe lesions (of magnesium deficiency ± vitamin D ± phosphate excess) resemble those of some congenital abnormalities. Lesions that develop later in infancy might provide the nidus for chronic cardiovascular and renal diseases of later childhood and adult life. Epidemiologic evidence is considered, having provided inferential evidence that magnesium deficiency (as in soft-water areas) contributes to the higher rate of sudden cardiac deaths (than in hard-water areas). Although differences in trace mineral and calcium contents of hard and soft water are also considered contributory, the most convincing evidence is that magnesium in hard-water areas is protective. Such a premise is subject to criticism because there are always concomitant factors that cloud the issue. Other dietary and environmental, as well as genetic, differences make it unlikely that there is a single provocative factor.
This book constitutes a plea for the objective examination of the evidence and for the exploration of the possibility that the prophylactic use of magnesium-especially in geographic areas where the intake is low, in families whose members have a high incidence of cardiovascular disease, and in high-risk individuals (e.g., diabetics and patients with a personal history of cardiac or vascular disease)-might be effective. Reevaluation of the use of vitamin D and of phosphate in foods is justifiable. The use of magnesium in the treatment of cardiac and renal diseases has been claimed by some investigators to be an important adjunct to therapy. More controlled studies should be done to obtain further evidence as to the extent to which experimental evidence and pilot clinical trials, indicative of benefits produced by magnesium, are applicable to more extensive treatment and prevention of human disease.
The substantial data on drugs (such as diuretics, cardiotonics, and antibiotics) that cause magnesium loss or inactivation are referred to only in the context of the theme of this volume and are so indexed. Further development will be provided elsewhere.
Appreciation is expressed to Harriet Nathan, May Becker, Marie Bennett, and Doris Wallace for typing the manuscript and to Dr. A. R. Berger for approving this employment of the secretarial staff of the Medical Service of Goldwater Memorial Hospital.
Mildred S. Seelig
New York
Contents
1 • Introduction: Consideration of Epidemiologic Factors
1.1. Ischemic Heart Disease
1.2. Concomitant Cardiovascular, Skeletal, and Renal Diseases
1.3. Changing Magnesium, Vitamin D, and Phosphate Intakes
1.4. Sex Difference in Magnesium Retention
1.5. Hard/Soft Water and Cardiovascular Disease
1.6. Epidemiologic Factors in Calcific Urinary Calculi
1.7. Genetic Factors in Cardiovascular, Skeletal, and Renal Diseases
(All figures and tables for Chapter 1)
Part I
Magnesium Deficiency during Gestation, Infancy, and Early Childhood
2 • The Role of Magnesium in Normal and Abnormal Pregnancy
2.1. Magnesium Balance in Pregnancy
2.2. Fetal Magnesium Requirements
2.3. Magnesium Serum Levels in Normal and Abnormal Pregnancy
2.3.1. Normal Pregnancy: Magnesium Levels
2.3.2. Preeclampsia and Eclampsia: Magnesium Levels and Treatment
2.3.2.1. Possible Contribution of Magnesium Deficiency to Eclamptic Pregnancy
2.3.2.2. Possible Contribution of Magnesium Deficiency to Placental and Coagulation Abnormalities
2.4. Magnesium Levels in Women with Recurrent or Imminent Abortion
(All figures and tables for Chapter 2)
3 • Consideration of Magnesium Deficiency in Perinatal Hormonal and Mineral Imbalances
3.1. Magnesium Deficiency during Gestation
3.1.1. Effects of Experimental Maternal Magnesium Deficiency on the Fetus
3.2. Perinatal Parathyroid Secretion: Interrelations with Magnesium and Calcium
3.2.1. Hyperparathyroidism of Pregnancy
3.2.2. Fetal Parathyroid Activity, and Phosphate, Calcium, and Magnesium Homeostasis
3.2.3. Hypoparathyroidism of Infancy
3.2.3.1. Hypocalcemia of Infancy
3.2.3.2. Magnesium Deficiency and Infantile Hypoparathyroidism
3.3. Calcitonin during Gestation; Interrelations with Magnesium and Calcium
3.3.1. Calcitonin during Pregnancy
3.3.2. Fetal Secretion of Calcitonin
3.3.3. Neonatal Calcitonin
3.4. Perinatal Hypervitaminosis D
3.4.1. Toxicity of Excess Vitamin D during Pregnancy
3.5. Summary of Maternal Factors That Might Contribute to Infantile Magnesium Abnormalities: Morbidity and Mortality
3.5.1. Genetic Hypoparathyroidism
3.5.2. Genetic Hyperparathyroidism
3.5.3. Reciprocal Maternal and Fetal Mineral Status
3.5.4. Maternal Age and Parity: Diabetes Mellitus
3.5.5. Eclampsia
(All figures and tables for Chapter 3)
4 • Magnesium Status in Infancy
4.1. Infantile Magnesium Deficiency: A Factor in Hypocalcemic Tetany, Seizures, and Respiratory Distress
4.1.1. Magnesium Deficiency in Metabolic Convulsions of Otherwise Normal Newborn Infants
4.1.2. Low-Birth-Weight Infants
4.1.3. Neonatal Hypoxia
4.1.4. Neonatal Infants of Diabetic Mothers
4.1.5. Neonatal Hypermagnesemia
4.1.6. Magnesium Depletion by Exchange Transfusions with Citrated Blood
4.1.7. Low Ionized Calcium and Hypomagnesemia
4.2. Treatment of Infantile Conditions Associated with Abnormalities of Magnesium
4.2.1. Correction of Neonatal Acidosis
4 2.2 Intensification of Magnesium Deficiency by Treatment of Hypocalcemia with Calcemic Agents
4.3 Influence of Infant Feeding on Magnesium Status Interrelations with Calcium, Phosphorus, and Vitamin D
4.3.1. Human versus Cows' Milk
4.3.1.1. Metabolic Balances of Infants Fed Human or Cows' Milk
4.3.1.2. Serum Magnesium, Calcium and Phosphorous Levels in Infants Fed Cows' and Human Milk
4.3.2. Risks of Excessive Vitamin D in Infancy
4.4. Primary Malabsorption of Magnesium
4.5. Acute and Protracted Gastroenteritis in Infancy and childhood
4.6. Protein Calorie Malnutrition (PCM)
4.7. Sudden Death in Infancy: Possible Role of Magnesium Deficiency
4.7.1. Sudden Infant Death Syndrome (SIDS)
4.7.1.1. Acute Magnesium Deficiency, Histamine Release, and Hypoxia in SIDS
4.7.1.2. Subacute Magnesium Deficiency and Cardiac Lesions in SIDS
4.7.1.3. SIDS and Hypoparathyroidism
4.7.1.4. Epidemiologic Factors in SIDS
(All figures and tables for Chapter 4)
Part II
Magnesium Deficiency in the Pathogenesis of Cardiovascular Diseases
5 • Failure to Reduce Incidence of Ischemic Heart Disease by Lowering Blood Lipids
5.1. Magnesium and Lipid Interrelationships
5.1.1. Influence of Fat on Magnesium Retention (Man)
5.1.1.1. Dietary Fat and Magnesium Balance
5.1.1.2. Steatorrhea and Magnesium Loss
5.1.1.3. Dietary Fat and Blood Lipids (Man)
5.1.1.4. Serum Magnesium and Cholesterol Levels in Cardiovascular Patients and High-Risk Populations
5.1.1.5. Clinical Use of Magnesium in Cardiovascular Disease with Hyperlipidemia
5.1.2. Blood and Cardiovascular Magnesium and Cholesterol in Experimental Dietary Atherogenesis and Cardiopathies
5.1.3. Magnesium/Lipid/Catecholamine Interrelationships
5.1.4. Estrogen, Lipids, and Magnesium; Interrelationships with Arteriosclerosis and Thrombosis
5.1.4.1. Estrogen Therapy of Ischemic Heart Disease
5.1.4.2. Estrogen, Cardiovascular Effects, and Magnesium
5.1.4.3. Magnesium, Estrogen, and Thrombotic Events
(All figures and tables for Chapter 5)
6 • Is Clinical Arteriosclerosis a Manifestation of Absolute or Conditioned Magnesium Deficiency?
6.1. The Arterial Wall and Arteriosclerosis
6.1.1. Mucopolysaccharides and Elastica in Arteriosclerotic Arteries
6.1.2. Pathology of Infantile Arteriosclerosis
6.1.3. Incidence of Infantile Coronary Arteriosclerosis
6.2. Factors Suggesting Magnesium Deficiency in Infantile Cardiovascular Disease
6.2.1. Experimental Arteriosclerosis of Magnesium Deficiency
6.2.1.1. Arterial Damage Caused by "Pure" Magnesium Deficiency
6.2.1.2. Arterial Damage of Magnesium Deficiency Intensified by High Calcium and Vitamin D Intakes
6.2.1.3. Arterial Damage of Magnesium Deficiency Intensified by High Fat Intakes
6.2.1.4. The Cardiovasopathic (CVP) Diet
6.2.1.5. Other Cardiovasopathic Models That Might Entail Relative Magnesium Deficiency
6.3. Catecholamine-Induced Arterial Damage; Magnesium Interrelationships
6.4. Magnesium Deficiency, Mast Cells, and Arteriosclerosis
6.5. Arterial Resistance, Blood Pressure, and Magnesium
6.5.1. Increased Arterial Resistance: Low Mg + K; High Ca + Na
6.5.2. Magnesium Deficiency and Decreased Blood Pressure; Refractoriness to Vasoactive Hormones
6.5.3. Clinical Magnesium Deficiency and Blood Pressure
(All figures and tables for Chapter 6)
7 • Magnesium Deficiency/Loss from Myocardium
7.1. Cardiac Magnesium Lability
7.2. The Magnesium Status of the Myocardium
7.3. Myocardial Changes with Magnesium Deficiency or Loss (Animal)
7.3.1. Experimental Magnesium Deficiency
7.3.2. Magnesium Loss from the Hypoxic Heart
7.3.3. Magnesium Loss from the Stressed Heart or in Association with Catecholamine Administration
7.3.4. Corticosteroid + Phosphate-Induced Myocardial Necrosis
7.3.5. Hereditary Cardiomyopathy of Hamsters
7.3.6. Stress and Free Fatty Acids/Myocardial Necrosis and Magnesium
7.3.7. Myocardial Loss of Magnesium after Parathyroidectomy and Sodium Phosphate Load
7.4. Cardiac Magnesium Loss: Central to Cardiac Dysionism, Disease, and Dysfunction
(All figures and tables for Chapter 7)
8 • Clinical Cardiac Abnormalities and Magnesium
8.1. Cardiomyopathies Not Secondary to Disease of the Major Coronary Arteries or to Infection
8.1.1. Peripartum Cardiomyopathy
8.1.2. Infantile Cardiomyopathy
8.1.3. Alcoholic Cardiomyopathy and Magnesium Deficiency
8.1.4. Diabetic Cardiomyopathy
(There are no figures and tables for Chapter 8)
9 • Magnesium Deficiency and Cardiac Dysrhythmia
9.1. Electrocardiographic Changes of Experimental Magnesium Deficiency
9.2. Magnesium Interrelationships with Other Factors in Cardiac Rhythmicity
9.2.1. Magnesium/Potassium in Cardiac Rhythmicity
9.2.2. Catecholamine/Magnesium/Potassium Interrelationships
9.2.3. Postinfarction/Catecholamine/Free Fatty Acid/Magnesium Interrelationships with Arrhythmia
9.2.4. Blood Primes for Extracorporeal Circulation
9.3. Magnesium Deficiency in Clinical Arrhythmia
9.3.1. Experimental Magnesium Deficiency (Man)
9.3.2. Electrocardiographic Changes with Use of ACD Blood
9.3.2.1. Exchange Transfusion
9.3.2.2. Open-Heart Surgery
9.3.2.3. Surgery, Drainage, and Magnesium-Free Intravenous Infusions
9.3.3. Malabsorption and Magnesium-Deficient Arrhythmias
9.3.4. Arrhythmias of Starvation
9.3.5. Arrhythmias of Alcoholism
9.3.6. Dysrhythmia in Diabetes Mellitus
9.3.7. Arrhythmias and Abnormal ECGs in Toxemias of Pregnancy and Peripartal Cardiomyopathy
9.3.8. Infantile Arrhythmias and Cardiomyopathies
9.3.9. "Idiopathic" and Postinfarct ECG Abnormalities That May Be Related to Magnesium Deficiency or Loss
9.3.9.1. "Benign" Arrhythmias
9.3.9.2. Similarity to ECGs of Magnesium Deficiency
9.3.10. Heart Block of Dialyzed Uremic Patients
(All figures and tables for Chapter 9)
10 • Therapeutic Use of Magnesium in Cardiovascular Disease
10.1. Magnesium in the Treatment of Arrhythmias
10.1.1. Magnesium and Digitalis Arrhythmias
10.1.2. Magnesium Treatment of Ischemic Arrhythmia
10.1.2.1. Magnesium in Experimental Hypoxic Arrhythmia
10.1.2.2. Magnesium in Clinical Arrhythmias of Ischemic and Unknown Origin
10.1.2.3. Glucose Solutions and Insulin to Increase Myocardial Magnesium and Potassium Uptake
10.1.2.4. The Role of the Anion
10.2. Formulation of a Metabolic Therapeutic Program for Treating Cardiomyopathies and Arrhythmias
(All figures and tables for Chapter 10)
Part III
Skeletal and Renal Effects of Magnesium Deficiency
11 • Magnesium, Bone Wasting, and Mineralization
11.1. Mobilization of Bone Magnesium
11.2. Influence of High Vitamin D and High or Low Calcium Intakes
11.2.1. High Calcium: Decreased Mobilization
11.2.2. Low Calcium: Increased Mobilization
11.3. High Phosphate Intakes: Effects on Bones
11.3.1. Effects on Bone Magnesium
11.3.2. High P/Ca; P/Mg and Bone Wasting; Mineralization
11.3.2.1. Bone Wasting
11.3.2.2. Bone Mineralization
11.4. Influence of Metabolic Activity of Bone on Availability of Bone Magnesium
11.5. Influence of Age on Mobilization of Bone Magnesium
11.6. Physicochemical Exchange of Bone Magnesium and Calcium
11.7. Alkaline and Pyrophosphatases, Magnesium, and Mineralization of Bone
11.7.1. Magnesium Requirement for Phosphatase Activation and Synthesis
11.7.2. Alkaline Phosphatase and Skeletal Mineralization (All figures and tables for Chapter 11)
12 • Abnormal Bone in Magnesium Deficiency
12.1. Osteopenia of Magnesium Deficiency (Animals)
12.2. Abnormal Bone: Hypermineralization and Hyperplasia of Magnesium Deficiency
12.3. Bone Diseases Possibly Related to Magnesium Deficiency
12.3.1. Fetal Magnesium Deficiency and Bone Damage
12.3.1.1. Interrelationships with Parathyroid Hormone and Calcitonin
12.3.1.2. Interrelationships with Gestational Hypervitaminosis D
12.3.2. Magnesium Deficiency and Bone Disease in Low-Birth-Weight Infants
12.4. Magnesium Status and Vitamin D Requirements and Responses
12.4.1. Increased Vitamin D Requirements of Magnesium Deficiency
12.4.2. Vitamin-D-Refractory Rickets and Osteomalacia
12.4.2.1. Hypophosphatemic Hyperparathyroid Rickets
12.4.2.2. Hyperphosphatemic Hypoparathyroid Osteopenia
12.4.3. Other Abnormal Function of, or Response to, Parathyroids
12.4.4. Osteopetrosis or Osteosclerosis and Hyperreactivity to Vitamin D
12.4.4.1. High Vitamin D and Calcium/Low Magnesium
12.4.4.2. Magnesium/Calcitonin Interrelationships in Osteoporosis
12.5. Other Genetic Bone Diseases and Possible Role of Magnesium
12.5.1. Osteogenesis Imperfecta
12.5.2 Hypophosphatasia
12.6. Other Osteopenias Possibly Mediated by Magnesium Deficiency
12.6.1. Osteoporosis
12.6.2 Renal Osteodystrophy
12.7. Joint Diseases Possibly Mediated by Magnesium Deficiency
12.7.1. Osteochondrosis
12.7.2. Chondrocalcinosis and Osteoarthritis
12.8. Magnesium Deficiency and Dental Disorders
13 • Renal Damage Caused by Magnesium Deficiency
13.1. Experimental Magnesium Deficiency
13.2. Intensification of Magnesium Deficiency Renal Damage by Excess Vitamin D (Animal)
13.3. Intensification of Magnesium Deficiency Renal Damage by Excess Phosphates (Animal)
13.4. Mediation by Secondary Hyperparathyroidism; Protection by Parathyroidectomy
13.5. Tissue Magnesium Loss and Damage: Not Parathyroid-Mediated
13.6. Phosphatases and Extraskeletal Mineralization
13.7. Magnesium Effect on Precipitation of Calcium Crystals in Urine
13.8. Clinical Renal Diseases Possibly Related to Magnesium Deficiency
13.8.1. Renal Tubular Defects in Magnesium Reabsorption
13.8.1.1. Contributions to Clinical Renal Magnesium Wastage by Calcemic Factors and Phosphate Therapy
13.8.1.2. Contribution to Clinical Renal Magnesium Wastage by Malabsorption
13.8.1.3. Miscellaneous Factors in Renal Magnesium Wastage
13.8.2. Renal Damage during Pregnancy: Related to Magnesium Deficiency?
13.8.3. Diabetic Renal Disease: Contributed to by Magnesium Deficiency
14 • Intensification of Magnesium Deficiency by Calcemic and Phosphate Therapy
14.1. Calcemic Therapy during Pregnancy
14.2. Calcemic Therapy during Infancy
14.3. Calcemic Therapy for Osteopenias
14.4. Treatment for Hypercalcemia
14.4.1. Risks of Phosphate Therapy
14.5. Complex of Diseases to Which Magnesium Deficiency Contributes Especially When Complicated by Calcemic and Phosphate Therapy
Appendix • Tests for Magnesium Deficiency
Cases of Infantile Ischemic Heart Disease
A.1. Limitations of Serum or Plasma Magnesium Levels
A.1.1. What is the Normal Range
A.1.2. Bound and Free Magnesium in Plasma
A.2. The Importance of Cellular Magnesium Determinations
A.2.1. Erythrocyte Magnesium
A.2.2. Skeletal Muscle Magnesium
A.2.3. White Blood Cell Magnesium Determinations
A.3. Percentage Retention of Parenteral Magnesium Loads
A.3.1. Recommended Procedures for Determining Percentage Retention of Parenteral Magnesium Load
A.3.1.1. Adults: Intramuscular Load
A.3.1.2. Adults: Intravenous Load
A.3.1.3. Infants: Intravenous Load
A.3.1.4. Infants: Intramuscular Load
A.3.2. Evaluation of Renal Handling of Magnesium
Bibliography (A-D)
Bibliography (E_K)
Bibliography (L_R)
Bibliography (S_Z)
================================================
Introduction: Chapter 1
The most alarming trend in the past half-century has been the sharp increase in sudden deaths from ischemic heart disease (IHD), particularly in middle-aged men, and the increasing number of younger men who suddenly develop myocardial infarctions, cardiac arrhythmias, or arrests. That men in the prime of life are thus afflicted is the dramatic and tragic tip of the iceberg. Underlying these catastrophes is the widespread increase in incidence of atherosclerosis in young age groups, and in myocardial hyperexcitability and cardiomyopathy without notable coronary atherosclerosis. It is proposed that magnesium deficiency or loss may be a common etiologic factor in the increased incidence of sudden infant deaths, infantile myocardial infarction and arteriosclerosis, and the disease that becomes manifest later in life. It is also suggested that magnesium deficiency might also cause or predispose to some skeletal and renal diseases, all of which can coexist.
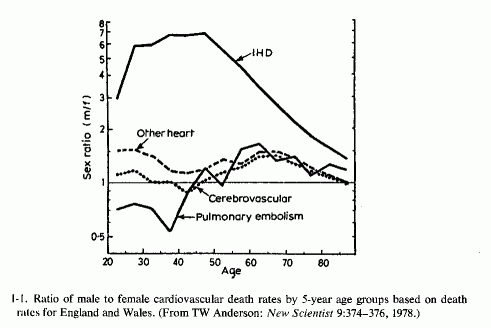
The cardiac problem in men has been deemed of sufficient magnitude as to be termed an epidemic that has been increasing, particularly since the middle 1930s. It has led to widespread institution of therapeutic and prophylactic regimens on the basis of suggestive findings. For example, young women have a significantly lower incidence of ischemic heart disease than do young men (Fig. 1-1). Because their α/β-lipoprotein ratios differ from those of the more susceptible young men and especially from those of patients with peripheral or coronary atherosclerosis, there was a period during which estrogens were widely used in the treatment of patients with myocardial infarctions and given prophylactically to high-risk (hyperlipidemic) men and postmenopausal women. This approach has been largely discontinued, predominantly because of the resultant increase in risk of thrombosis Another approach that was given a trial period was administration of excesses of unsaturated fatty acids; the incidence of atherosclerosis and IHD is lower in countries where more vegetable oils than saturated animal fats are consumed. A modification of the fatty-acid-supplement regimens that has been receiving extensive clinical trial is to replace saturated with unsaturated fats. This approach has lowered blood lipids, but not the incidence of IHD. Because altering fat intakes of patients with established hyperlipidemia and atherosclerosis has not reduced the mortality from IHD, it has been recommended that the time to institute such a dietary modification might be in early infancy, a suggestion that has been disputed.
{kind=link}
Among women, the incidence of atherosclerosis and IHD increases with age, especially after the menopause, often in association with osteopenia or with calcific renal disease. The combined problem of bone wasting and extraskeletal calcification (particularly renal and cardiovascular) is also encountered in renal osteodystrophy and in other conditions associated with hyperparathyroidism and phosphate treatment of hypercalcemia.
Rarer forms of osteopenia, usually found in association with cardiac anomalies, arteriosclerosis, and renal calcinosis, are seen in infants of low birth weight, or who have ontogenesis imperfecta or hypophosphatasia. The more common, but not widely known, arteriosclerosis and IHD of early infancy is also usually accompanied by renal calcinosis, as is the later form that is accompanied by hyperlipidemia, hypertension and atherosclerosis. The latter type-some forms of which are associated with aortic and pulmonary stenoses and atresias, and with endocardial fibroelastosis-has been attributed to hypervitaminosis D (Seelig, 1969b; Seelig and Haddy, 1976/1980) which contributes to loss of magnesium. These conditions are stressed in this volume because they support the supposition that atherosclerosis (and some renal and skeletal diseases) have their roots early in infancy and have put the onus on the absolute or conditioned magnesium deficiency that has become a problem during this century.
Magnesium plays an important role in maintaining the integrity of the myocardium, kidneys, and bone. Its deficiency has been shown to cause cardiomyopathy in several animal species, and to intensify myocardial lesions caused by a variety of modalities. Its deficiency has caused arteriosclerosis and has intensified formation of atheromata, or arteriosclerosis, thrombosis, and even myocardial infarction, induced by atherogenic diets, high intakes of vitamin D, calcium, phosphate, and fat. Its deficiency has caused renal lesions and intensified damage produced by vitamin D, calcium, and phosphate. And its deficiency has been implicated in some forms of bone damage. Magnesium supplementation has prevented or reversed some of the lesions in the experimental models and been used clinically in cardiovascular disease and urolithiasis.
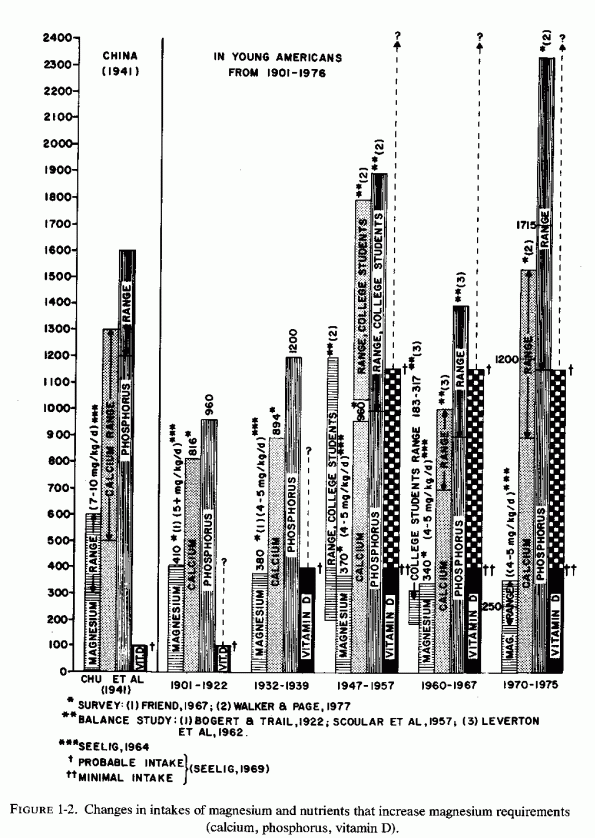
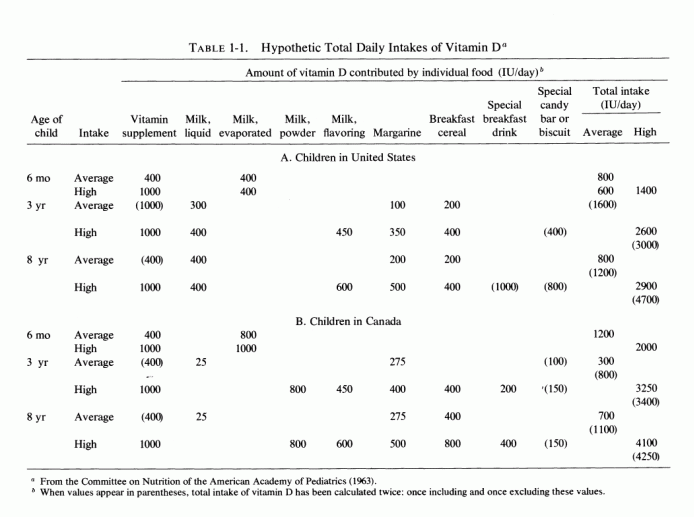
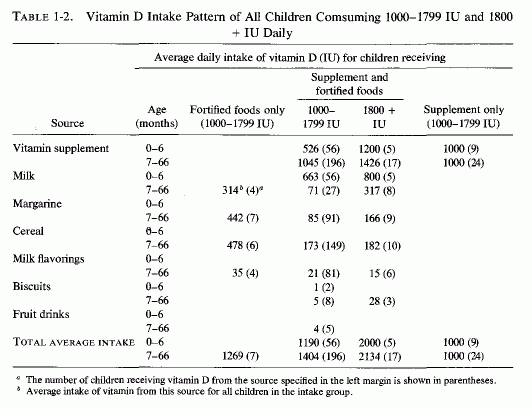
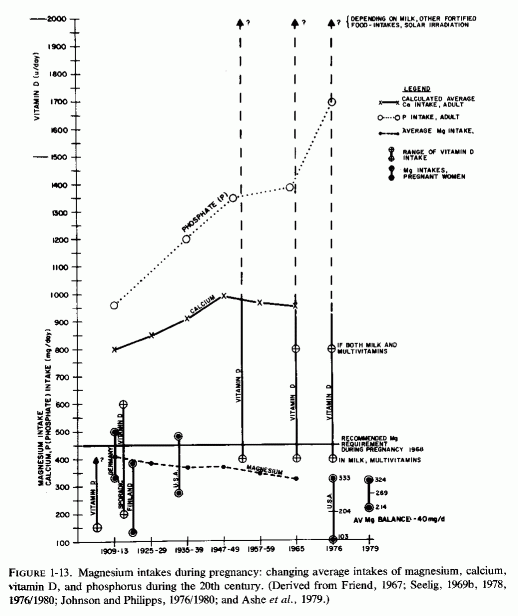
Examination of the changing nutritional intakes in America, particularly from the middle 1930s is disconcerting in light of these experimental findings. Although magnesium intakes have been gradually falling since the beginning of the century, there were sharply increased intakes of nutrients that increased its requirements [particularly high vitamin D and phosphorus intakes (Seelig, 1964, 1971) subsequently (Fig. 1-2)]. The rise in vitamin D intake began when the addition to each quart of milk of a sufficient amount (400 IU) to cure, rather than merely to prevent, rickets became widespread from the mid 1930s and was made mandatory in most states from the 1940s to 1950, either replacing cod liver oil, or taken in addition to it (Baldwin, 1953; Seelig, 1969b, 1970b). Fortification of many foods in addition to milk, including milk flavoring, oleomargarine, breakfast cereals, or "substitutes," led the Committee on Nutrition of the American Academy of Pediatrics to express concern about the total daily intake of vitamin D in the United States, which they calculated might range from 600 to 4000 IU/day from marketed fortified products (Table 1-1). A survey of 1000 Canadian children from 1 week to 51/2 years of age showed that 70% consumed more than 400 IU, and 30% consumed over 1000 IU of vitamin D daily (Broadfoot et al., 1972). Table 1-2 depicts the sources of vitamin D among those receiving over 1000 to 1800 IU of vitamin D per day. The major source of phosphorus derives from soft drinks that contain phosphoric acid, the consumption of which has been rising markedly in the last quarter of a century (Henderson, 1972; Lutwak 1974).
{kind=link}
{kind=link}
{kind=link}
Although it is generally believed that the rise in blood lipids is due to increased intakes of saturated fats during this century, and that sugar consumption has also increased substantially, comparison of per capita intakes from 1909 to 1965 shows relatively minor changes (Fig. 1-3). The average daily fat intake rose from 112 to 132, but most of the increase has been in unsaturated fatty acids. The total carbohydrate intake dropped from 492 to 374, so that the greater percentage increase of sugar in 1965 reflects an increase of about 40 grams daily. Probably the sugar intake has risen more since the 1965 value (Fig. 1-2) among those who drink larger quantities of sugar-sweetened, phosphorus-containing soft drinks.
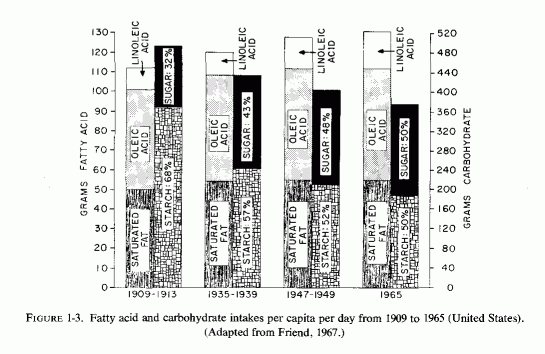{kind=link}
Largely disregarded is the possibility that the hyperlipidemia associated with atherosclerosis might be caused by hypervitaminosis D, which also causes hypertension (Linden, 1977; Seelig and Haddy, 1976/1980), as well as the more widely recognized complications; cardiovascular and renal damage, and hypercalcemia (Seelig, 1969b). Much of the clinical data on the cardiovascular, skeletal, and renal damage caused by vitamin D derives from the use of massive doses of vitamin D a quarter of a century ago in the treatment of such diseases as rheumatoid arthritis, and from the lesser overdosage of European children at a time when administration of up to 4000 IU/day was not uncommon (Table 1-3; Seelig, 1969b). The sharp rise in vitamin D intake depicted for the 1947-1957 segment of Fig. 1-1 is presumed because of the probable consumption of large quantities of milk by the college students studied-an impression suggested by their high calcium intake (Scoular et al., 1957), in contrast to the lower intake noted in a general diet survey (Friend, 1967). Since the amount of vitamin D needed by most adults is considered so small as to be met by exposure to sunlight and by ingestion of natural (unfortified) foods (Food and Nutrition Board, 1968), such high intakes must be considered well into the toxic range. As long ago as 1932, L. Harris reported that in the human, the toxic dose of vitamin D is not far removed from the therapeutic (antiricketic) dose. Stewart (1964) reported that there is a narrow toxic-therapeutic ratio. Furthermore, even most infants are protected against rickets by as little as 100 IU of vitamin D daily (Fraser, 1967), whereas a survey of young Americans showed that 50% ingested 400-800 IU daily, 10% usually consumed over 1000 IU daily, and occasionally as much as 2900 IU were taken (Dale and Lowenberg, 1967). Epidemiologic data have correlated moderately high vitamin D intake with increased incidence of myocardial infarction, renal calcinosis, and urolithiasis (Linden, 1974a,b). In northern Norway, where intake of natural foods rich in vitamin D is common, the incidence of hypercholesterolemia and susceptibility to sudden death from ischemic heart disease and to calcific renal diseases, two conditions which are often found in the same patient (Linden, 1972, 1975/1977; Westlund, 1973), seems to be related to the amount of vitamin D ingested and to the individual sensitivity to solar irradiation. Since magnesium deficiency is also associated with abnormal lipid distribution, and vitamin D excess causes magnesium loss, interrelations of protracted high intakes of vitamin D with magnesium requirements, and with the cardiovascular and renal lesions of each imbalance, deserve study (Seelig, 1977).
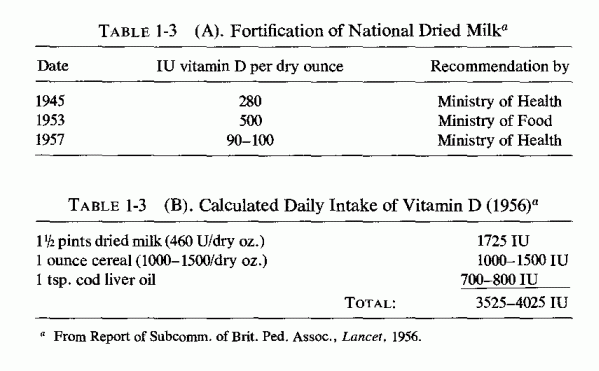{kind=link}
Like magnesium deficiency and hypervitaminosis D, excess phosphate has also been implicated in cardiovascular, skeletal, and renal damage. The nature of the pathologic changes produced by dietary excesses of phosphorus depends upon its ratios to both calcium and magnesium. Figure 1-2 shows that the phosphorus intake increased sharply in the college studies during the periods analyzed in 1947-1957 (Scoular et al. 1957), and in the most recent survey of college diets (Walker and Page, 1977). The lower phosphorus level entered in the 1960-1967 block of columns derives from an extensive metabolic balance study in several colleges (Leverton et al., 1962). One can speculate that during these strictly controlled periods there was likely to have been less consumption of soft drinks containing phosphoric acid than during the self-selected dietary intakes reflected in the college diet surveys.
The recommended phosphorus/calcium (P/Ca) ratio is 1.5/1 (U.S. Department of Agriculture Report, 1972). In 1932-1939, the P/Ca ratio was about 1.2/1; it was estimated to be rising to as much as 4/1 among those who substitute sodas for milk (Lutwak, 1974). This shift in ratios was stressed as potentially harmful to bones, as a result of secondary hyperparathyroidism, on the basis of the effect of the osteopenia produced by comparable P/Ca dietary ratios in several species of animals, up to the monkey (Krook and Barrett, 1962; Krook et al., 1963, 1971; Henrikson et al., 1970; Draper et al., 1972; Krishnarao and Draper, 1972; Krook et al., 1975).
However, the most recent dietary survey of college diets from fifty colleges (M. Walker and Page, 1977) showed that the mean P/Ca ratio was about 1.5/1, both phosphorus and calcium intakes having risen to 1200 and 1700 mg/day, respectively. What had dropped was the magnesium intake-to a mean of 250 mg/day. Such diets provide dietary ratios of Ca/Mg and P/Mg of almost 5/1 and almost 7/1, respectively. Since an excess of either phosphorus or calcium has been shown to increase magnesium requirements and to intensify signs of magnesium deficiency (Reviews: Seelig, 1964, 1971), such a dietary pattern-particularly when accompanied by high vitamin D and phosphate intakes by many-can be expected to produce either absolute or relative magnesium deficiency.
Analysis of published metabolic balance studies (such as are done to establish a nutritional requirement, an amount sufficient to maintain equilibrium) has shown that young men require more magnesium in mg/kg/day than do young women (Fig. 1-4) (Seelig, 1964). The studies analyzed had been obtained from throughout the world, and showed that young Americans tended to ingest less magnesium on self- selected diets than did Orientals and, on average, tended to be in negative balance. This was particularly so for the young men, who on the average excreted more magnesium than they ingested on the typical American intake of 4-4.9 mg/kg/day. Young women on that typical intake, on the other hand, tended to remain in equilibrium. The typical magnesium intake of the Orientals studied was between 7 and 10 mg/kg/day, and positive balance or equilibrium was the rule. In deriving the recommended magnesium intake from the data analyzed, the intake was selected at which equilibrium or positive balance was reached in at least three-fourths of the subjects. On this basis, the minimal daily requirement is 6 mg/kg/day. For a 140-lb woman, this comes to 385 mg of magnesium daily; for a 185-lb man, at least 500 mg/ day. Americans, and others in industrialized countries, tend to ingest diets rich in other nutrients (fat, protein, sugar, phosphorus, and vitamin D), all of which increase magnesium requirements (Seelig, 1964, 1971; Lindeman, 1976/1980). In addition, moderate to heavy ingestion of alcohol (even as "social" drinking) is not uncommon, and alcohol is magnesuretic (McCollister et al., 1958, 1963; Kalbfleisch et al., 1963). Thus, a magnesium intake of 7-10 mg/kg/day might be preferable. On this basis, a 185-lb man might require 580-800 mg/day of magnesium, probably approximately twice as much as his diet normally delivers. Possibly a woman (unless she is pregnant or lactating) requires somewhat less. The most recent survey of college students (from 50 colleges) shows that less than the modest officially recommended amount [300 mg for women; 350 mg for men (Food and Nutrition Board, 1974)] is the amount usually ingested (M. Walker and Page, 1977). Actually, the mean daily magnesium intake of the college students (250 mg) may well be no more than half the amount required by the young women; it may be as little as one-half to one-third the amount needed by large, athletic young men. In contrast to their inadequate magnesium intake, they ingest one and a half times the recommended amount of calcium and twice the phosphorus allowance. Consumption by young college women of diets that provide suboptimal amounts of magnesium is not unique to the 50 colleges surveyed. N. Johnson and Philipps (1976/1980) surveyed the diets of pregnant women from different economic brackets, and found that their magnesium intakes ranged from 103 to 333 mg/day, with an average of 204 mg daily, an amount grossly inadequate for pregnant women. Ashe (1979) confirmed the inadequacy of prenatal magnesium intakes of 10 healthy white women from private practices in Tennessee by 7-day metabolic balance studies done at intervals throughout pregnancy. Their mean daily magnesium intakes were only 60% the recommended 450 mg/day, and mean balances were -40 mg/day. Only 3 of the 47 periods were positive. The investigators suggested that high calcium, phosphorus, and protein intakes might have intensified the severity of the negative magnesium balances. The significance of such low magnesium intakes during gestation, as regards the cardiovascular, skeletal, and renal status of infants of women with gestational magnesium deficiency, is considered in Part I of this volume.
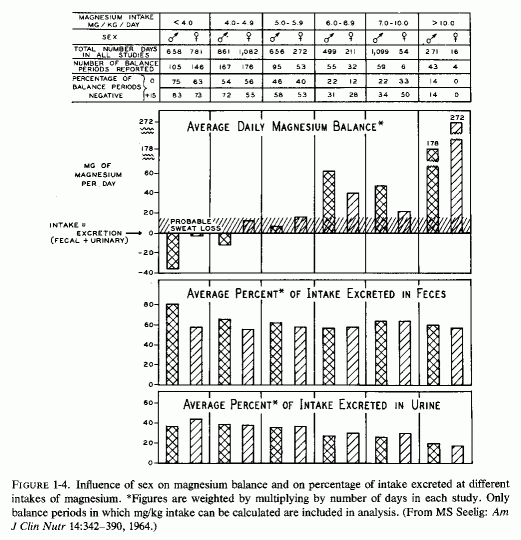{kind=link}
Now that high fiber- (and phytate-) containing diets are increasingly being recommended, the effect of such diets on a magnesium intake that is otherwise meager should be explored. Review of metabolic studies of magnesium utilization by subjects on diets rich in phytates-brown bread, brown rice, oatmeal, or white bread to which phytate had been added-showed poor percentage absorption of the magnesium, particularly when the diet was first changed (Seelig, 1964). After several weeks on the phytate-rich diet, the absorption of magnesium tended to improve (A. Walker et al., 1948; Cullumbine et al., 1950; Hathaway, 1962). McCance and Widdowson (1942a,b) found that addition of phytate to white bread caused greater fecal magnesium excretion, and removing phytate from brown bread greatly improved magnesium absorption. Reinhold et al. (1976) have recently confirmed these observations, not only for magnesium but for trace metals. Thus, the higher magnesium content of phytate-containing whole grain products may not be a reliable source, in terms of availability of magnesium. Whether adaptation to the phytate ingested, on its continued inclusion in the diet, will result in better utilization (as suggested in the early cited studies) remains to be investigated systematically.
Long-term metabolic studies provide a more valuable index of adequacy of intake than do short-term studies. Figure 1-5 shows that on very low intakes (< 4 mg/kg/day) the young men remained in negative balance for the average of 52 days of study, whereas the young women retained sufficient magnesium at the end of their 30 days to maintain equilibrium, even taking into account probable sweat loss. On the usual American intake of 4-4.9 mg/kg/day, the young men went into equilibrium at the end of the study; the young women were in magnesium balance throughout. Why there was less magnesium retention by the young men whose intakes were slightly higher (5-5.9 mg/kg/day) is puzzling. Perhaps that group happened to have higher intakes of nutrients that interfere with magnesium absorption or increased renal magnesium excretion. Continuation of strong positive balances after a month on supplements that raised the magnesium intakes of young men to 9.7-12.7 mg/kg/day suggests restoration of a deficit. A subsequent study by Irwin and Feeley (1967) showed sustained strongly negative magnesium balances (-77, -74, and -38 mg/day) in 15 healthy women evaluated for 3 consecutive 20-day periods that delivered 230-300 mg of magnesium daily. They concluded that the recommended daily intake of magnesium (300 mg) is insufficient to maintain magnesium equilibrium in 140-lb women, and suggested that the proposed intake of 385 mg/day (Seelig, 1964) might be a preferable amount. In a long-term study (50 and 20 weeks) of 3 men on magnesium intakes of 1 .8 mg/kg/day to 5 mg/kg/day, Tipton and Stuart (1970) found that the young man who weighed 100 kg who was on the diet delivering the least magnesium (180 mg/day) or 1.8 mg/kg/day lost an average of 90 mg of magnesium daily during the 50-week study. A smaller (71 kg) young man given twice as much magnesium (that provided 5 mg/kg/day) retained an average of 70 mg/kg/day. An 85-kg middle-aged man who was fed a diet containing 310 mg of magnesium daily (3.8 mg/kg/day) lost an average of 40 mg daily during the 20 weeks on study. In a long-term study of men (in a Veterans Administration Hospital Metabolic Unit) Spencer et al. (1976/1980) found that increasing the magnesium intake about fourfold over the amount supplied (about 250 mg) in the basic diet did not consistently increase the amount of magnesium retained. About two-thirds of the supplement was excreted in the feces. The amount of calcium and phosphorus in the diet and the duration of the metabolic periods influenced the results. On low to high daily calcium intakes, magnesium-supplemented (about 500 mg/day patients retained about 49 to 58 mg of magnesium daily on low calcium intakes (200 mg daily). Patients on 1400-mg calcium intakes remained in slightly negative magnesium balance (-8 mg/day) when they were supplemented with magnesium; when they were not given the extra magnesium their daily magnesium loss was 20 mg. Adding the magnesium supplement to a diet plus calcium supplements providing 2000 mg of calcium raised the magnesium balance from + 2 to + 85 mg/day. Increasing the phosphorus intake to close to 1500 mg from 975, converted a positive magnesium balance (+29) to a negative one (-19 mg/day) during a period of low calcium intake, but not when the calcium intake was also increased. Spencer et al. (1979) suggested that the different amounts of magnesium retained by the different supplemented patients might have reflected their prior magnesium status. This impression is supported by the high retentions of magnesium by supplemented subjects who had previously been subjected to magnesium deprivation (Fitzgerald and Fourman, 1956; Shils, 1964, 1969a,b). They (Spencer et al. 1976/1980) also stressed the importance of the duration of the study, noting that, during the early phase of their studies, the positive magnesium balances were strong; several weeks later, the patients were in equilibrium or even in slightly negative balance. Perhaps this reflects repletion of an insufficiency, such as had been postulated might occur with sufficiently sustained magnesium supplementation (Seelig, 1964).
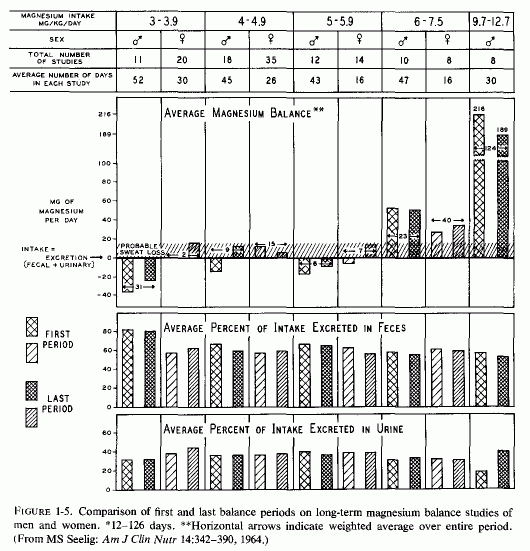{kind=link}
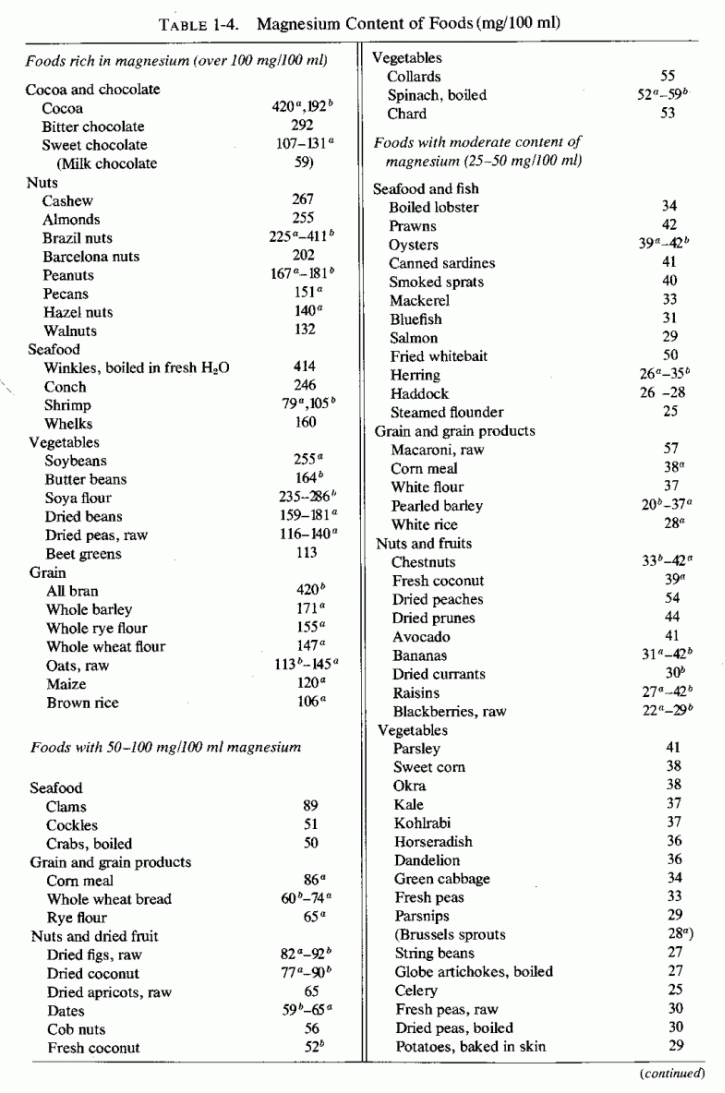
The cited dietary surveys and metabolic balance studies support the contention that magnesium supplied by the American diet-and most likely by that of most industrialized countries, particularly those populated by Europeans or by those with comparable eating habits-is likely not to be optimal. Such intakes, which are at best marginal, can be frankly deficient when there are concomitant high intakes of nutrients that increase magnesium requirements. Manifestly, although the incidence of abnormalities that resemble those produced in experimental or conditioned magnesium deficiency has increased during the years that the dietary pattern has changed to one that leads to at least conditioned magnesium deficiency, such abnormalities are not found in the entire population. Individual (or familial or group) differences in dietary habits can be partially responsible. (Table 1-4 gives magnesium content of foods.) Also probably contributory are genetic differences in utilization or retention of magnesium and in vitamin D metabolism (Seelig, 1969b, 1970a,b). It is hoped that future investigation will resolve whether the familial instances of parathyroid dysfunction and of some congenital cardiovascular or renal diseases are related to basic genetic variants in the handling of magnesium and vitamin D, and whether those two recognized genetic variants are interrelated.
{kind=link}
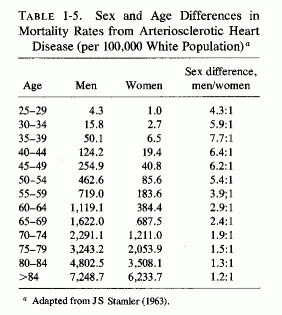
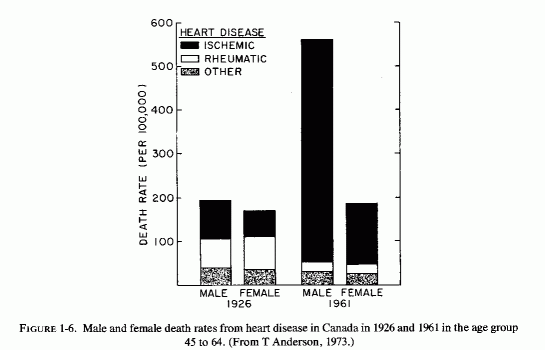
One wonders whether the demonstrated better retention of magnesium by women than men on marginal magnesium intakes can contribute to the dramatic sex difference in incidence of IHD in young adults (Table 1-5; Figure 1-1) and to the rise of incidence in death rates in Canada from 1926 to 1961 (Fig. 1-6, T. Anderson, 1973). The sharp increase that occurred only in middle-aged men was entirely in the IHD category; cardiac death rates from other causes dropped. Among the women, the cardiac death rate remained the same, but the proportion due to IHD rose. There was a lesser sex difference in the proportion of deaths that occurred suddenly in the middle-aged groups in hard- and soft-water cities in Ontario, and still less in the 65 to 74 year-old groups (T. Anderson et al.1976/1980). Whether the observation of this group that myocardial magnesium levels were lower in women who had died suddenly (accident or suicide) than they were in comparable men, both in hard- and soft-water areas (T. Anderson et al., 1978), bears on this question requires resolution. On the surface it would seem to militate against the concept that women's better retention of magnesium explains the sex difference in the rise of IHD. Additional factors must be considered. Among such factors are those diagrammed by Raab (1972), who had earlier provided experimental evidence that stress causes decreased myocardial magnesium levels (Raab et al. 1968). Does this imply that women are more subject to stress-induced decreased myocardial magnesium? This seems dubious. More likely, women normally have less myocardial magnesium than do men. Does the amount of muscular exertion play a role? The higher myocardial magnesium levels in left than in right ventricles (Holtmeier, l969a; Szelenyi, 1973) might be germane to this point.
{kind=link}
{kind=link}
The evidence that dietary magnesium is generally insufficient and that under those conditions women retain more than do men, is clear, however-wherever the magnesium goes. It provides some insight into the provocative epidemiologic studies that demonstrate that the cardiovascular death rates are higher in areas supplied with soft water than they are in hard water areas. N. Goldsmith (1969) and Hankin et al. (1970) have calculated that 12% of the daily intake of magnesium can be derived from water. Among those using only hard water, as much as 18% of the daily magnesium intake may derive from water. Among those whose magnesium intakes from food are marginal, these amounts might well be critical.
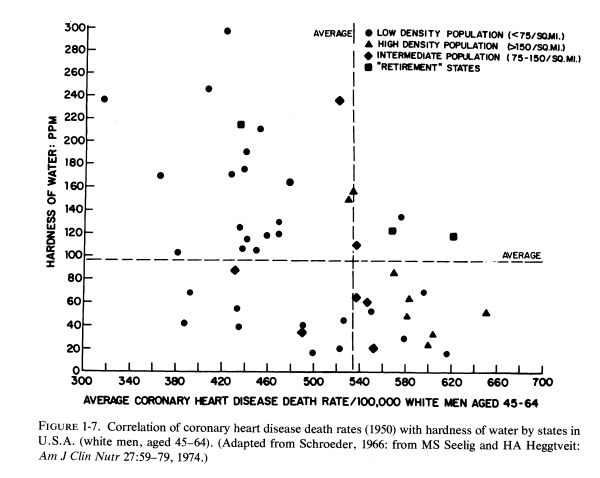
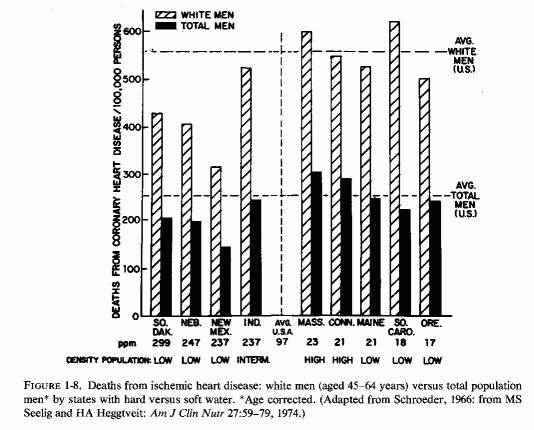
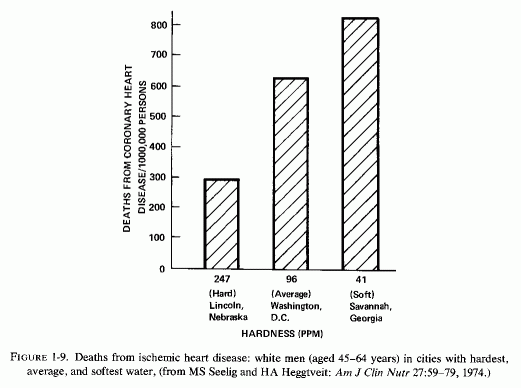
J. Kobayashi (1957) first noted that the nature of drinking water might influence death rates from cardiovascular disease; the incidence of strokes is high in areas with acid (soft) water. Schroeder (1960a,b, 1966) surveyed the hardness of drinking water in each of the United States, and correlated the death rates with state-wide water hardness or softness (Fig. 1-7). He found that death rates from cardiovascular diseases (particularly from "coronary" heart attacks in white men 45-64 years old) were significantly higher in states with soft water than in states with hard water (Fig. 1-8). The death rate in South Carolina, a state with the softest water, was 983/100,000; that in Nebraska, a hard-water state, was 712/100,000. Deaths from cerebrovascular accidents followed a similar pattern. Complicating interpretation of these findings is the fact that ischemic heart disease death rates are higher in urban than in rural communities. To eliminate this factor, the coronary death rates from three cities with hard-, intermediate-, and soft-water supplies are compared (Fig. 1- 9), and reveal a startling contrast between the rates of fatal ischemic heart disease in cities with hard and soft water. Since this observation, there have been many confirmatory studies, although there has not been complete accord that it is the magnesium, rather than the calcium, that is protective, or whether there might be a toxic element in the soft water that is to blame, e.g., cadmium (Perry, 1973), copper (Harman, 1975/1977), or others (Editorial, Lancet, 1969c).On the other hand, Klevay (1975, 1977, 1978) has presented provocative evidence that a high zinc/copper dietary ratio might contribute to ischemic heart disease as a result of relative copper deficiency, which causes a decrease in high-density lipids and an increase in low-density lipids. He has suggested that hard water might be protective by lowering the ratio of zinc to copper. Where hard water is artificially softened (i.e., by sodium chelates), the role of increased sodium should also be considered.
{kind=link}
{kind=link}
{kind=link}
M. Crawford et al. (1968) like others working in areas where calcium is by far the major water factor (J. Morris et al., 1962; Biorck et al., 1965; J. Robertson, 1968, 1969), favored calcium as the probable protective factor. In England, the average calcium content of hard water accounts for about 84% of the hardness and is more than 11 times greater than the magnesium content (M. Crawford et al. 1968). Nonetheless, it was her impression that the water "factor" probably involves interrelationships of the "bulk" ions: calcium, magnesium, and sodium (M. Crawford, 1972). Those who had had favorable experience with the use of magnesium salts in treating patients with acute or chronic IHD (R. Parsons et al., 1961; Berberian, 1962; Browne, 1961, 1963, 1964a,b) favored magnesium as the hard-water protective factor. So also did those who had done or evaluated animal work that showed magnesium to be protective and excess calcium either not protective or harmful in experimental cardiomyopathies or soft-tissue calcification (e.g., cardiovascular and renal) (Neal and Neal, 1962; Marier, 1963; Bajusz, 1967; Marier et al., 1968; Seelig and Bunce, 1972; Seelig and Heggtveit, 1974). In his consideration of the part played by hard water, Bajusz (1967) suggested that the higher content of magnesium might protect the myocardial cell against damage caused by ischemia and improve its ability to resist the effects of cardiotoxic agents. In 1974, Seelig and Heggtveit considered the experimental and clinical evidence that calcium and magnesium exert reciprocal effects on myocardial irritability. High calcium levels stimulate and high magnesium levels suppress hyperexcitability. They then suggested that magnesium might be useful in maintaining normal cardiac rhythmicity, in the face of ischemia or digitalis or in acute (i.e., alcohol- or diuretic-induced) hypomagnesemia [The antiarrhythmic attribute of magnesium is again being utilized in the United States (Chadda et al., 1973a,b, 1976/1980; Iseri et al., 1975; Iseri and Bures, 1976/1980) after a hiatus of 30 years (Boyd and Scherf, 1943).]
The dietary surveys presented here show that magnesium, but not calcium, intakes have been gradually falling. Coinciding in time with the sharp increase, first of vitamin D and then of phosphorus intakes (Fig. 1-1), there has been a steep increase in incidence of sudden deaths from IHD (T. Anderson and LeRiche, 1970). The recognition of this increase in IHD death rates derived from an extensive study of death certificates of men 45-64 years of age (Ontario, 1901-1961). As many as 5000 certificates a year had to be examined when the incidence was low (in 1901). Where deaths from IHD were clearly specified, as compared with all cardiac deaths, it was the IHD category that had risen, more than doubling from 1931 to 1961 (Anderson and LeRiche, 1970). The death rates from other major forms of heart disease in that age group had fallen during the same period of time. The minor changes in cardiac death rates from 1901 to 1931 are not as readily interpreted, because of changes in terminology and possible incompleteness of reporting. Selecting 1931 as the earliest key date (sudden-death coroner reports being available from about 1931 on Toronto), these investigators found that only about half of the non- traumatic sudden deaths were attributed to IHD in 1931, whereas 99% were deemed due to IHD in 1961. Spain et al. (1960), on the basis of an autopsy survey, considered such events the commonest cause of death of middle-aged men, at about the same time. T. Anderson et al. (1969) postulated that there might be an environmental factor that could, by altering myocardial excitability, cause an increase in the incidence of sudden death from ventricular fibrillation and other cardiac arrhythmias, and noted that the sudden death rate (but not the nonsudden IHD death rate) varies inversely with the hardness of the water. T. Crawford and M. D. Crawford 1967), who had noted that despite a much higher frequency of cardiac death rates in Glasgow (a soft-water area) than in London (a hard-water area) degrees of coronary atherosclerosis were not dissimilar, had also suggested that the water factor might affect the way the myocardium reacts to ischemia. They found that the coronary magnesium content was higher in young men (under 40) who had died as a result of accidents in London than in Glasgow, and that the Scottish young men had more small myocardial scars than did the Londoners.
From Ontario, where the magnesium content of hard water is much higher than it is in England, has come much of the definitive data implicating magnesium rather than calcium as the protective factor in hard water, and ruling out most of the potentially toxic trace minerals found in soft water as the harmful soft-water factor. In their surveys of cardiac death rates, T. Anderson et al. (1969) found that there were many more (sudden) cardiac deaths reported in soft- than in hard-water areas (T. Anderson et al., 1978). This supported T. Crawford and M. D. Crawford's (1967) and Bajusz's (1967) suggestion that the hard-water factor was likely to be a myocardial protective factor. They speculated that it probably affected cardiac rhythmicity (T. Anderson et al., 1969, 1973, 1976/1979; T. Anderson and LeRiche, 1970, 1971). Comparable findings were reported by Fodor et al. (1973) from Newfoundland, where there is a much higher death rate for IHD in a city with very soft water than in two communities with hard water, particularly for men, 62% of whom died before they could be brought to the hospital (considered probable sudden deaths). They commented that IHD death rate in men in the soft-water city (702/100,000) is comparable to that in the "high mortality belt" of the southeastern portion of the United States.
At first Anderson et al. (1969) adhered to the English premise that calcium was likely to be the protective water factor. When they became aware of the evidence that Western diets provided marginal amounts of magnesium (Seelig, 1964) and that persons dying of heart attacks have low myocardial magnesium levels, even in non-infarcted segments (Heggtveit et al., 1969; Seelig, 1972), they had pathologists from hard- and soft-water areas secure myocardial specimens from routine autopsies, and had them analyzed for magnesium, calcium, and trace elements (T. Anderson et al., 1973, 1975, 1976. Magnesium was the only element with a significant difference in myocardial concentration, which was higher in hearts of accident victims from hard-water areas (982/918 µ/g dry tissue). IHD disease victims had 22% lower myocardial magnesium levels in soft- than in hard-water areas (697/744). In England, there has been an apparently contradictory pattern (Chipperfield et al., 1976a), with lower levels of myocardial magnesium in hard- than in soft-water cities. T. Anderson et al. (1978) point out that since in the two English cities that were compared the magnesium levels are quite low both in the hard and in the soft water (Chipperfield et al., 1976b), the difference between them represents only 1% or 2% of the probable total intake, and that another factor might be operative.
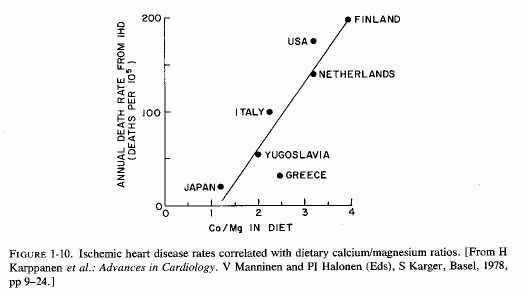
In Finland, which has a very high death rate from IHD, there is a clear relationship with the amount of magnesium in the soil (Karppanen and Neuvonen, 1973). In eastern and in northern Finland, where the soil content is about a third that found in southwestern Finland (Karppanen et al., 1978) the mortality from ischemic heart disease is twice as high as is that in the southwest. Ho and Khun (1976/1980) surveyed factors that might be contributory both to the rising incidence of cardiovascular disease in Europe, and the falling levels of magnesium both in the soil and in the food supply. They commented that in Finland, which has the highest cardiovascular death rate in Europe, the dietary supply of magnesium had decreased by 1963 to a third of the intake common in 1911 (H. Katz, 1973). In contrast, in Japan with its low cardiac death rate, the daily magnesium intake was cited as 560 mg (Holtmeier, 1969a, 1973). Karppanen et al. (1978) have depicted the steep rise in ischemic heart disease that coincides with increasing dietary calcium/magnesium ratios (Fig. 1-10).
{kind=link}
In view of the possibility that sudden deaths of infants might similarly be mediated by magnesium deficiency, and be analogous to the adult cardiac arrhythmic sudden deaths that are prevalent in soft-water areas, the preliminary report by Godwin and Brown (1973) of a somewhat higher incidence of sudden infant deaths in soft-water counties in California than in hard-water counties is provocative. It must be noted that the following year a conflicting report was published (Allwright et al., 1974) that failed to confirm the higher incidence of either IHD of adults or of infant mortality rates with soft water. However, these investigators point out that this "soft" water is approximately as hard as is the "hard" water in some of the English studies, where higher infant-death rates were reported in soft- than in hard-water areas (M. Crawford et al., 1972). These tentative findings call to mind the instance of sudden infant death that was associated with coronary arteriosclerosis (Meurman et al., 1965) from eastern Finland, and the report by Pesonen et al. (1975) on more severe and more frequent infantile coronary arteriosclerosis in eastern Finnish children than in those from the southwest (where the magnesium level is higher).
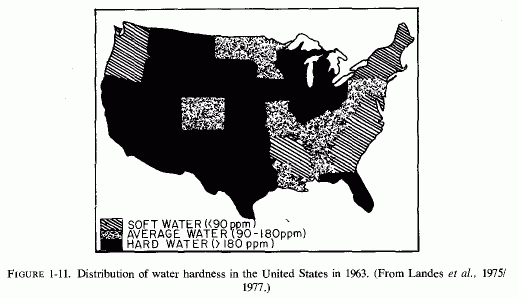
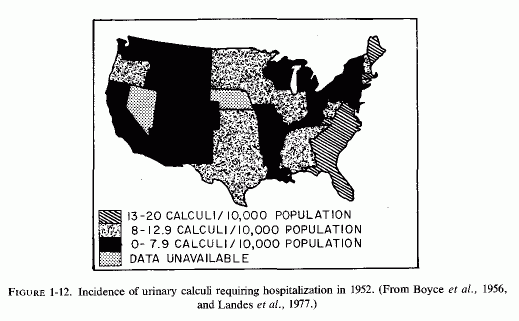
There has been an increase, during this century, in the incidence of calcium oxalate stones in Finland (Sallinen, 1960), central Europe and Sweden (Grossman, 1938; Hedenberg, 1951) and Japan (Inadaet al., 1958; Editorial, Brit Med J, 1965) that coincides with the rising incidence of cardiovascular diseases in those countries, and with the use of magnesium-poor soil fertilizers in the northern and central parts of the European continent (Holtmeier and Kuhn, 1976/1980). The geographic difference in frequency of calcific urolithiasis in the United States (Landes, 1975/1977; Finlayson, 1974; Landeset al., 1977) coincides with the geographic difference in incidence of sudden death from ischemic heart disease, and with the degree of water hardness. A map of the United States, indicating the distribution of water hardness in 1963 (Fig. 1-11) and one showing the incidence of urinary calculi(Fig. 1-12) clearly shows that in most states where the water is softest the frequency of urolithiasis is highest. Boyce et al. (1956), who pointed out this geographic overlap, reported that the highest incidences of kidney stones were in South Carolina and other southeastern states, an area that has been called "the kidney stone belt," and the lowest incidences were in midwest and southwestern states, where the water is hard. Melnick et al. (1971, 1973) and Landes et al. (1977) reaffirmed this observation, basing their conclusions on hospital diagnoses, obtained from a survey done in 1972. South Carolina again came in first, with the highest frequency (17/1000 discharges). Nebraska, the state shown earlier to have the lowest incidence of sudden death from ischemic heart disease, also had the lowest frequency of urinary calculi (2.6/1000 hospital discharges). Accepting the limitations of such state-wide surveys of stone incidence and water quality, the authors nonetheless felt justified in concluding that the differences were statistically significant, indicating that the incidence of urinary calculi is inversely related to the hardness of the water (Landes et al. 1977).
{kind=link}
{kind=link}
Prien (1971), who had reported that magnesium therapy in conjunction with pyridoxine (Prien, 1965; Gershoff and Prien, 1967), was useful as prophylactic therapy for recurrent calcium oxalate stone-formers (in northern New England, another soft-water area), presents "the riddle of urinary stone disease." He referred to Grossmann's (1938) evidence that, starting in 1924, the incidence of small calcific stones in young adults rose in central and northern Europe, and was puzzled as to why the incidence should have dropped during World War II, only to rise again thereafter (Boshamer, 1961). It is possible that the work of Linden (1972, 1974a, 1977) correlating concurrent urolithiasis, hyperlipidemia, and ischemic heart disease with only moderately high intakes of vitamin D might be germane to the rise in incidence of kidney stones after 1924. Linden (1977) mentioned that after Mellanby (1920) had demonstrated that cod liver oil could prevent rickets; it was soon widely used for lesser ailments, such as failure to thrive and poor appetite. He referred to reports, in the late 1920s, of infantile fatalities due to hypervitaminosis D. It is possible that inappropriate and widespread use of vitamin D, which increases magnesium requirements, might have intensified magnesium deficiency, the predisposition for which might have derived from the decreased magnesium-availability from the soil, especially in those parts of central Europe where fertilizers high in potassium and low in magnesium were commonly used after World War I (Aleksandrowicz and Stachura, 1976/1980; Holtmeier and Kuhn, 1976/1980). Perhaps, during World War I the vitamin D supplements and soil fertilization were less widely used, only to be taken up again after the war. In the last year of World War I and for more than a decade thereafter, in the British Isles, excessive vitamin D was provided in infant formulas and other foods, with a resultant epidemic of supravalvular aortic stenosis syndrome (SASS) and increased incidences of renal tubular acidosis, infantile nephrocalcinosis, and osteosclerosis. In Germany, "Stosstherapie" with huge parenteral doses of vitamin D also caused SASS and related "congenital" abnormalities (Review: Seelig, 1969b). To what extent the long-term use of therapeutic doses of vitamin D in infants and children with low requirements or hyperreactivity to vitamin D (Seelig, 1970a,b), and to what extent its continued use throughout life, and especially during adolescence and early adulthood when milk consumption tends to be high (in America), might predispose to a high urinary calcium/magnesium ratio and to a conditioned magnesium deficiency should be systematically investigated. Possibly it might be part of the answer to Prien's kidney-stone riddle (1971), as well as to the continued high sudden-cardiac death rate.
The inverse relationship between the tendency toward calcium oxalate urinary tract stones and the tendency toward osteoporosis (McGeown and Oreopolis, 1969) is provocative. There are fragmentary data indicating that magnesium deficiency contributes to several pediatric osteopenias and to osteoporosis, all of which are characterized by loss of matrix. A high Ca ratio might favor hypermineralization of bone with defective matrix. High P/Ca and P/Mg ratios might favor osteomalacia. Correlation of these mineral ratios with hormonal responses might shed some light on the high rate of osteoporosis in postmenopausal women, who might have a high parathyroid/estrogen ratio, in addition to loss of the estrogen-induced capacity to store magnesium in bone Whether low magnesium and high vitamin D intakes during pregnancy contribute to osteogenesis imperfecta, hypophosphatasia, and fragile bones of low-birth-weight infants should be studied.
Even though the dietary factors (high vitamin D and phosphate intakes and declining magnesium intakes) have been widespread, and in the case of vitamin D unavoidable for milk-drinkers, the increased incidence of frequency of some cardiovascular, skeletal, and renal diseases has not been distributed equally in the population. Except for osteoporosis, which is most prevalent in white postmenopausal women (McGeown, 1969; Meema et al., 1973, 1975; N. Goldsmith and Johnston, 1978), and hypertension, which is most prevalent in black women (Kuller et al., 1973), most of the diseases for which evidence is presented in this volume, of relationships to low magnesium, to vitamin D, and to phosphate intake, are most prevalent in white males. Furthermore, there is evidence of familial predisposition to what may be risk factors: (1) specific magnesium malabsorption and renal wasting, and (2) hyperreactivity to vitamin D (Seelig, 1969b, 1970a,b). It is suggested that the familial instances of calcium oxalate urolithiasis (McGeown, 1960; Resnick et al., 1968), of pseudohypoparathyroidism with vitamin-D-resistant rickets (DeLuca et al., 1967; Falls et al., 1968; Reitz and Weinstein, 1973), and possibly of hyperparathyroidism (Cutler et al., 1964; Cholod et al., 1970; Marx et al., 1973) might also be secondary to a primary abnormality in magnesium metabolism, leading to magnesium deficiency. Several forms of neonatal or infantile cardiovascular disease, possibly including juvenile hyperlipidemia and hypertension, might also be related to abnormalities in magnesium or vitamin D metabolism, or both, as might vitamin-D-resistant rickets. The familial instances of these diseases and of other osteopenias, which are not infrequently associated with renal disease, might have an underlying defect: magnesium malabsorption or renal tubular wastage or both.
The renal and skeletal disorders contribute to significant morbidity. The cardiovascular complications lead both to morbidity and sudden mortality. An editorial (JAMA, 1972) entitled "A Magic Carpet Is Not Enough" calls urgent attention to the fact that as many as over 50% to 73% of sudden deaths from lethal arrhythmias (Kuller et al. 1967; Armstrong et al., 1972) occur before the patients reach the hospital. Among almost a thousand cases of medically untreated deaths from IHD in which autopsies were done, 60% of the men and 47% of the women died within 15 minutes of onset of symptoms (Wikland, 1971). The reference to the inadequacy of the "magic carpet" pertained to the finding that even if the patient is resuscitated, death is usually merely somewhat delayed by a period of invalidism (Geddes et al., 1967). In a confirmatory study, Kuller et al. (1973) showed that 75% of those who died suddenly had had no serious disability; only 12% had been unable to work. Among those who died in a hospital, only 17% survived more than 24 hours. Of the almost 500 who died within 24 hours of onset of symptoms who were autopsied, only 13% had evidence of a recent infarct. The investigators concluded that their findings indicated that no current community health approach will be effective. They state that a fundamental change in therapeutic and preventive approach is needed.
The correlation of data implicating interrelationships between absolute magnesium deficiency and the magnesium-losing excesses of vitamin D and phosphates in so many of the diseases that have increased in incidence during the time that these dietary imbalances developed might point toward the new approach that Kuller et al. (1973) said was required. Such imbalances are a particular risk during pregnancy (Fig. 1-13). Possibly they might lead to abnormal pregnancy, to congenital abnormalities, and to infantile and later morbidity and mortality.
{kind=link}
Now that vitamin D has been proved to have a steroid hormone mode of action (Norman, 1968; Norman et al., 1975/1977; DeLuca, 1969, 1976), and the active antirachitic metabolites have been isolated, synthesized, and made available, vitamin-D-resistant rickets can be treated specifically, in preference to putting an entire population at risk of hypervitaminosis D by fortifying milk and other foods (Seelig and Mazlen, 1977). Perhaps the popular soft drinks that provide so much of the excess phosphate can be reformulated to be bubbly by other means than by use of phosphate salts. And, finally, physicians should evaluate their patients, particularly those with the cited familial disorders-for abnormal magnesium metabolism, and they should prescribe supplements when needed.
This volume places major emphasis on the early establishment of cardiovascular, skeletal, and renal lesions-possibly during gestation, infancy, and early childhood-that can either cause early manifestations of disease or death, or lay the groundwork for disease processes that become overt later in life. Thus, the first part deals with prenatal, perinatal, and infantile disorders, and the subsequent parts deal more generally with the abnormalities of the three systems to which magnesium deficiency might well be contributory.
Part I: Chapter 2
MAGNESIUM DEFICIENCY DURING GESTATION, INFANCY, AND EARLY CHILDHOOD
The formation of new tissue (maternal and fetal) during pregnancy requires higher magnesium intakes than that of the normal nonpregnant woman of comparable age. The most recent recommended dietary allowances in the United States and Canada is 450 mg/day (Food and Nutrition Boards, 1968), a figure that is probably based largely on magnesium balance determinations and calculations done with adult pregnant women from 1914-1942. The general statement that the dietary magnesium during pregnancy should substantially exceed the amount required by other adults has led to the selection of 450 mg/day as reasonable, exceeding that recommended for adolescent and young adult women in the United States by 100 mg/day and exceeding the amount recommended in Canada for women over 22 by 150 mg/ day. Since adolescent children require much higher magnesium intakes to meet their own growth and maturation needs, it is questionable whether the same amount deemed necessary for the mature pregnant woman is sufficient for a teenaged pregnant girl. Even the amount generally considered sufficient, but rarely met by the American woman, whether or not she is pregnant (Seelig, 1964; N. Johnson and Phillips 1976/1980; Ashe et al., 1979), should be reevaluated.
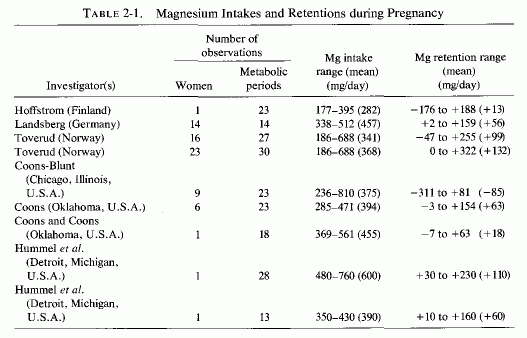
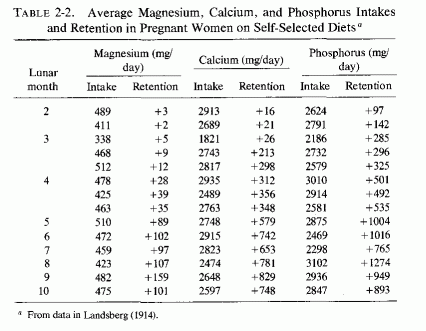
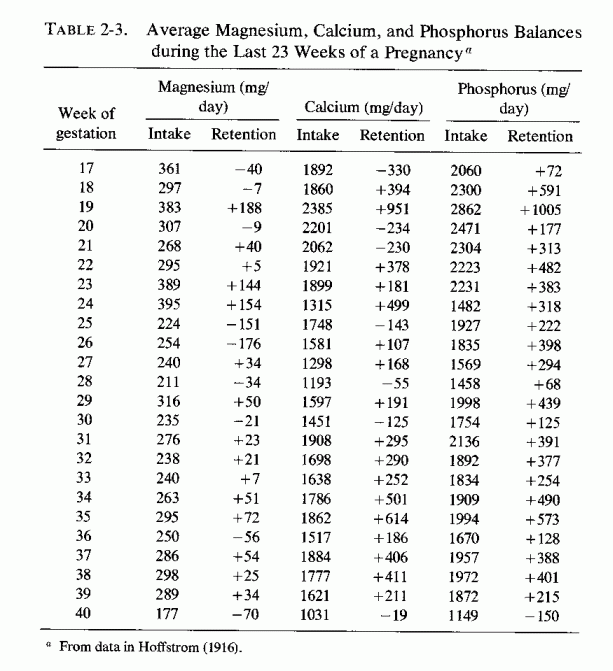
Examination of magnesium retention by pregnant women on different dietary intakes (Table 2-1, Seelig, 1971) shows marked differences in retentions, ranging from negative to strongly positive. The first detailed metabolic balance studies of pregnant women (in Germany) that gave magnesium, calcium, and phosphorus intakes and retentions (Table 2-2, Landsberg, 1914) showed strongly positive balances of all these elements. The magnesium contents of the self-selected diets of 14 women ranged from 338-512 mg/day, and their calcium and phosphorus intakes were usually between 2 and close to 3 g a day. Hoffstrom's long-term study of a Finnish pregnant woman's metabolic balances during the last 23 weeks of pregnancy (Table 2-3, Hoffstrom, 1916) showed that on her much lower magnesium intakes, she was in negative magnesium balance during nine of the periods and retained less than 50 mg/day in eight more. Despite her adequate calcium and phosphorus intakes in all but four periods (never falling below 1 daily) she was in negative calcium balance during seven periods. She rarely retained as much calcium or phosphorus as did the women in the German study (Landsberg, 1914).
{kind=link}
{kind=link}
{kind=link}
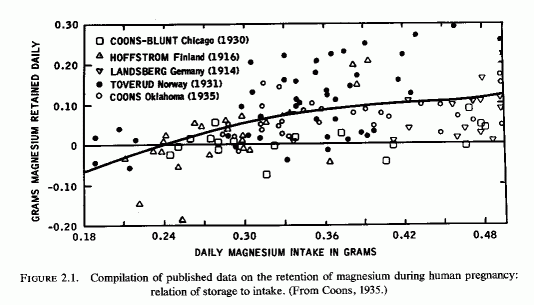
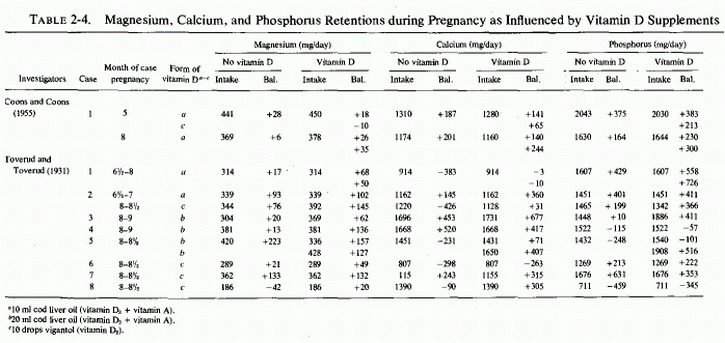
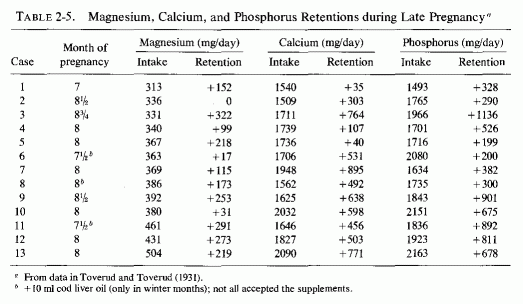
The emphasis in the United States was largely on the problem of calcium retention, and Coons and Blunt (1930) at first studied magnesium balances of pregnant women to see whether taking milk of magnesia as a laxative would unfavorably influence calcium retention. They found no interference with calcium retention, even on magnesium intakes as high as 810 mg/day. Toward the end of pregnancy, there was a tendency toward more and larger negative magnesium balances, even on daily magnesium intakes of 400 mg/day. They subsequently compared their findings with those obtained by other investigators (Fig. 2-1, Coons, 1935). The composite curve, and the scatter diagrams, show weakly positive and even negative magnesium balances on daily intakes of less than 300 mg/day. In their own study of eight women in Chicago (Coons and Blunt, 1930), half of the metabolic balance periods showed net losses of magnesium. There was a preponderance of positive balances in their Oklahoma studies of six women (Coons et al. . 1934, 1935); they speculated that the greater exposure to sunlight in Oklahoma might have been responsible for the better magnesium retention in their 1935 studies. To test the possibility that vitamin D was responsible, they studied the effect of cod liver oil on the magnesium retention of a primiparous woman who had also been tested before pregnancy (Coons and Coons, 1935), and whose intakes of magnesium and phosphorus were kept fairly constant. This was a long-term investigation that included 18 metabolic periods of 4 days each on a continuously regulated diet, from the 21st to 30th weeks of pregnancy. Despite an apparently adequate intake of magnesium (369-561 mg/day), three negative balances occurred during three of the metabolic periods, and the woman's average daily retention of magnesium was only 18 mg. Exposure to sunshine was avoided and cod liver oil supplements were provided only during 25th, 26th, 34th, and 35th weeks of study. The investigators concluded, from the slightly lower calcium and magnesium retentions during the first two weeks of cod liver oil administration at five months gestation, and the minimal changes in calcium retention and slight increase in magnesium retention during the second two weeks of supplementation during the eighth month of gestation, that vitamin D from cod liver oil was not equivalent (in its effects on calcium and magnesium retention) to that from reasonable exposure to sunlight (Coons and Coons, 1935). Table 2-4 includes the above data, and the balance data from the study of Toverud and Toverud (1931), from women whose mineral intakes were kept fairly constant before and while on vitamin D supplementation. The Norwegian study (Toverud and Toverud, 1931) shows that the magnesium balances improved on addition of vitamin D supplements, even when the magnesium intake was low (case 8). In that instance, the vitamin D converted a negative calcium balance on an adequate calcium intake to positive, but did little to correct the negative phosphorus balance, the phosphorus intake also being low. The women whose calcium and magnesium intakes were fairly low, but whose phosphorus intakes were adequate (cases 1,6), responded to vitamin D with more retention of magnesium, much less negative calcium balance in one (case 1) but no significant diminution of the strongly negative calcium balance in the other (case 6), whose phosphorus balances remained strongly positive. Not included in this table are the data from women given diets with and without added calcium as salt and milk, which showed that they required at least 1.6 g of calcium and phosphorus daily to maintain positive balances of those elements. The effects of the increased intake on magnesium retention cannot be determined from that study because the magnesium intake was not constant. In a subsequent study, in which the daily dietary intakes of calcium and phosphorus were kept at 1.5-2 g and that of magnesium between 313 to 504 mg (Table 2-5), the three women whose magnesium intakes exceeded 430 mg/day all obtained strongly positive magnesium balances. The one with the highest intake, whose intakes of calcium and phosphorus were over 2 g, retained slightly less magnesium than did those with slightly lower calcium and phosphorus intakes. Another woman whose magnesium/calcium/ phosphorus intakes were 392/1625/1843 also showed high retentions of all three elements. One with comparable magnesium intake (380) but calcium and phosphorus intakes above 2 g retained only 31 mg of magnesium daily. There are exceptions to these findings; individual differences and variations in intakes of effective elements no doubt influenced the metabolic balances. These data are suggestive that the dietary ratios of magnesium, calcium, and phosphorus, and a requisite amount of vitamin D, all influence the retention of these elements during pregnancy.
{kind=link}
{kind=link}
{kind=link}
The long-term studies of a 37-year-old multiparous woman with a history of three prior successful pregnancies and healthy babies (Table 2-6, Hummel et al. 1936), and of an 18-year-old primipara with a suboptimal nutritional background but on a good diet during pregnancy (Table 2-7, Hummel et al., 1937), provide some data that might be germane to the lower magnesium levels of young primiparas and of their infants at birth. The healthy woman, whose metabolic studies encompassed 28 metabolic balance periods from the 135th to 280th day of pregnancy, was on an unusually rich diet that included two quarts of milk, each of which contained 400 units of vitamin D as cod liver oil. This provided an excess of calcium and phosphorus over that considered desirable, and exceeded that shown by Toverud and Toverud (1931) to decrease the retention of magnesium to +31 mg/day in the woman (case 10, Table 2-5) receiving 380 mg magnesium daily, but not to decrease its retention in the woman (case 13, Table 2-5) who ingested about 500 mg of magnesium daily. Neither received vitamin D supplements. Similarly, the patient reported by Hummel et al. (1936, Table 2-6) had high average daily magnesium intakes of 590-615 mg/day during the last two months of pregnancy, the month in which Toverud and Toverud did their metabolic studies (Table 2-5, 2-6), and then retained an average daily amount of magnesium of 85-104 mg. The poorly nourished primipara whose metabolic balance determinations were performed from 60 to 5 days antepartum (the length of gestation was not specified) exhibited greater daily calcium retention and lesser daily magnesium retentions during most of the metabolic balance periods. Only during two of the periods did she retain more than100 mg of magnesium daily. Calculations of the retention of the well-nourished quadripara during the 65 days up to 5 days before delivery, to obtain figures comparable to those for the 65-day period during which the young primipara was studied, show that the total gains during the last two months of pregnancy up to five days before birth were:
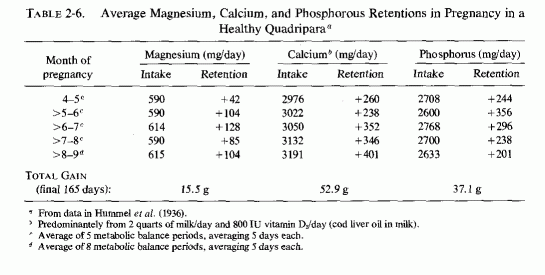{kind=link}
{kind=link}
| Element (g) | Primipara | Quadripara |
| Magnesium | 4.2 | 8.0 |
| Calcium | 46.3 | 25.3 |
| Phosphorus | 16.3 | 12.7 |
Provocative is the finding that the primipara retained about half as much magnesium and almost twice as much calcium as did the healthy thirty-seven-year-old mother of three healthy children. The greater magnesium retention of the older woman is readily understandable on the basis of her having regularly ingested almost 200 mg more magnesium daily than did the young girl. Her lesser retention of calcium is surprising in view of her having regularly ingested extremely high amounts of calcium (about 3 g daily), in contrast to the acceptable intakes of close to 2 g daily by the young girl.
The magnesium intake of the woman who had had successful pregnancies and healthy offspring (Hummel et al., 1936) is reminiscent of the early metabolic studies by Landsberg (1914). In both, all of the metabolic balance determinations showed retentions of magnesium, as well as of calcium and phosphorus. Since Landsberg's 1914 study in Germany, analysis of self-selected diets of pregnant women have shown that daily intakes of magnesium ranged from 260 mg to below 400 mg in 9 out of the 12 studies evaluated (Coons and Coons, 1935). Two subjects ingested 413-422 mg daily; only one selected a diet that delivered 500 mg/day. The calcium and phosphorus intakes were usually close to the recommended amounts. A recent study of 47 pregnant women residing in Wisconsin (N. Johnson and Phillips, 1976/1980) showed that their daily intake was even less adequate than had been cited in the 1935 study. Their magnesium intakes ranged from 103-333 mg/day, averaging 204 mg± 54 S.D. daily. None ingested the recommended 450 mg/day; 98% ingested less than 70% of the recommended daily allowance; and 79% ingested less than 55%. The lower magnesium intakes were correlated with low birth weights. Ashe et al. (1979) have recently shown similarly low intakes in middleclass pregnant women. They had an average daily loss of 40 mg of magnesium.
Coons et al. (1935) tabulated the mineral constituents of fetuses by lunar month, obtained from the literature. Table 2-8 provides their magnesium, calcium, and phosphorus data. It should be kept in mind that human fetuses available for such analyses are usually obtained as a result of abnormalities during pregnancy or labor. Thus, their constituents cannot be considered indicative of those of normal fetuses or full-term infants. As an example, among the analyses by Givens and Macy (1933) were twins born after eight lunar months: one died in three hours and had a total magnesium content of 670 mg; the other died after four days and had a total magnesium content of 1443 mg, far more than might be retained in those few days. Magnesium balance data tabulated for newborn infants (Duckworth and Warnock, 1942), suggest total daily retentions of magnesium of 10-18 mg). Thus, the mineral contents of fetuses and neonates have a wide range at any given age, possibly reflecting maternal stores and intake and placental integrity. Widdowson and Spray (1951) analyzed the mineral content of fetuses, tabulating the data by body weight. The data on magnesium, calcium, and phosphorus are given in Table 2-9. The increments of minerals reflect both the growth and changing chemical composition of the fetus as it develops. Widdowson and Dickerson (1962) have illustrated the changes by comparing the composition of a fetus weighing 175 g with its composition at 3.5 kg, were its chemical composition to be increased proportionally twentyfold, and the actual composition of a 3.5-kg infant (Table 2-10). Infants born prematurely have considerably less of these minerals than do full-term infants, with relatively lesser amounts of calcium and phosphorus than of magnesium, indicating the lesser bone calcification, most of which occurs in the third trimester. The magnesium content of neonates has been as low as 277 mg and as high as 886 mg; similarly, the calcium content of the newborn has been from 13.08 to 33.27 g, and that of phosphorus, 8.96-18.68 g (Coons et al.1935).
{kind=link}
{kind=link}
{kind=link}
When the dietary intake of magnesium is not sufficient to meet the demands of gestation, the maternal stores are mobilized and magnesium deficiency can develop. Although under most circumstances the body maintains plasma magnesium levels within very narrow limits, the pregnant woman tends to develop lower than normal magnesium levels, even in the absence of toxemia. Since the homeostasis of calcium and phosphorus is intimately related to that of magnesium, brief note is taken here of the tendency also toward declining calcium levels during pregnancy (Newman, 1957; Hardy, 1956; E. Dawson et al., 1969; Watney et al., 1971). It has been shown that phosphorus levels also fall somewhat during pregnancy, so calcium supplements have often been given in the form of the phosphate, with resultant increase in leg cramps of pregnancy. Hardy (1956) and Kerr (et al. (1962) demonstrated that when the phosphate salt is given, with or without vitamin D (viosterol), the serum total and ionized calcium levels were actually depressed, as compared with the rises seen in pregnant women given calcium carbonate or lactate. Even the serum phosphate levels increased when the nonphosphate calcium salts were given (Kerr et al., 1962). Since high phosphate intake interferes with magnesium, as well as calcium absorption, it is possible that calcium phosphate salts also lowered magnesium levels, and that this might have contributed to the muscle cramps.
The first reports of blood magnesium levels during pregnancy were in 1923. Krebs and Briggs (1923) reported a range of 1.7-2.2 mEq/liter among 17 women in their 8th to 40th weeks of pregnancy. Bogert and Plass (1923) compared the serum levels of 40 pregnant women at different stages of pregnancy with those of nonpregnant women and found that the average value 2.0 mEq/liter at the outset (which equaled the control average) fell to an average of 1.7 by the end of pregnancy. Watchorn and McCance (1932) found that half of the 12 pregnant women in their series had serum magnesium levels below 1.99 mEq/liter (which was below the values they found in normal nonpregnant subjects), and that the percentage of the total magnesium in the ultrafiltrable fraction was increased. They were dubious that the difference was due to diminished quantities of serum protein, this not being a constant finding, and speculated that an unidentified change in the physicochemical equilibria must have taken place that allowed for more ready passage of magnesium across the placental barrier. Such a change might allow, too, for more ready urinary excretion and might partially explain the need for high magnesium intake during pregnancy to maintain the degree of positive balance necessary for successful gestation without prejudicing the health of the mother. Another group of investigators reported that the blood magnesium of 75 women was higher especially in the sixth month of gestation (range during pregnancy 1.95-2.78 mEq/liter mean = 2.41) than it was four nonpregnant women (2.11 mEq/liter) (Zaharescu-Karaman et al., 1936a). However, they found that the level dropped markedly at the end of labor, to a range of 0.35-2.35 mEq/liter and a mean of 1.5 (Zaharescu-Karaman et al., l936b). Extremely low serum magnesium levels (1.0-1.1 mEq/liter) were reported in a small series of cases by Wolff and Jorrand Bourquard (1937) in the second month of pregnancy which increased slightly (to 1.25-1.41 mEq/liter) at the end of gestation. Their control (nonpregnant) mean value was 1.7 mEq/liter. Haury and Cantarow (1942) included four normal pregnant women in their tabulation of 108 subjects, and reported a range of 1.4-2.1 mEq/liter; most of their normal controls had serum magnesium levels of 1.8-2.4 mEq/liter. Köberlin and Mischel (1958) also reported lower Mg levels in the first trimester than later in pregnancy. A more extensive report by Newman (1957) has shown the range of serum magnesium levels in 27 normal pregnant women to be very wide in each of the trimesters, at delivery, and at 3-5 days and 6 weeks postpartum (Table 2-11). Newman also reported an unusually wide normal range of serum magnesium (1.34-2.4) in non- pregnant women. The calcium and phosphorus levels also dropped slightly.
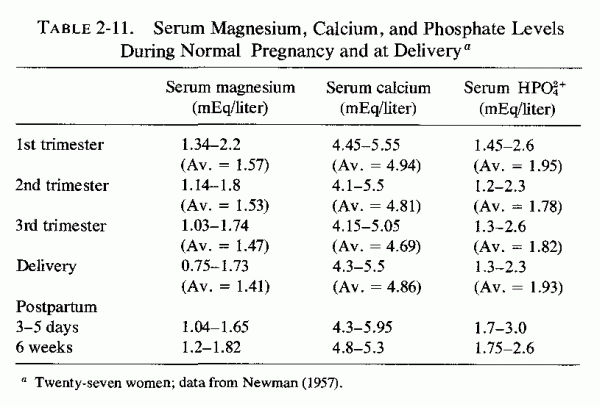{kind=link}
Hall (1957) graphed values, obtained from 30 pregnant patients who were followed from 11 weeks to term and at six weeks postpartum (Fig. 2-2), as well as values from 294 normal and toxemic (11.9%) women. Their work illustrates that the normal pregnant woman tends to have serum magnesium levels that remained at the low limit for the nonpregnant range (1.69-2.0 mEq/liter) with the broadest range (about 1.6-2.1) in the second trimester. The lowest values that were recorded in this study, which started in the 11th week, were in the 12th-18th week of pregnancy (1.45-1.8 mEq/liter. Archari et al. (1961) found no difference in serum magnesium levels of normal pregnant and nonpregnant women; both groups had a range of 1.5-1.9 mEq/liter. DeJorge (1965a,b) found that serum magnesium levels fell continuously in 99 pregnant women from about 1.6 mEq/liter in the second month to about 1.2 mEq/liter in the eighth month, as compared to their nonpregnant range of 1.70-2.25 mEq/liter. Correcting for the dilution of plasma that occurs during pregnancy, in a study of 139 pregnant women (Table 2-12), they concluded that the hypomagnesemia is real only during the first half of pregnancy and during the last month (DeJorge, 1965b). Comparable conclusions were reached by Dawson et al. (1969) in their study of 244 adolescent (ages 13-19) pregnant women. The mean plasma magnesium levels declined slightly from 2.6-2.2 mEq/liter as pregnancy progressed, but showed no change when expressed as a ratio to hematocrit values.
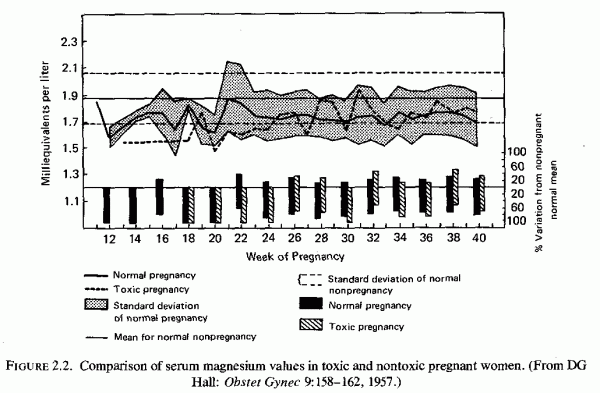{kind=link}
{kind=link}
Celli Arcella (1965) reported lower serum magnesium levels (1.9 mEq/liter) during the third trimester than in normal nonpregnant women (2.2 mEq/liter). Lim et al. (1969b) similarly reported significantly lower serum (and erythrocyte) levels in normal pregnant women in the third trimester than in normal nonpregnant women. In the latter study, the average of 105 serum samples from normal pregnant women was 1.43 ± 0.05 mEq/liter, with a range of 1.28-1.73, as compared to the normal nonpregnant value of 1.60 ± 0.17. The average value for erythrocyte Mg was also lower than for nonpregnant women. The authors suggest that these differences, taking into account the increasing demands of the rapidly growing fetus, may indicate an occult magnesium deficiency. In contrast, Mahran and Hanna (1968) reported a higher mean (1.83 ± S.D = 0.28) among normal pregnant women in the third trimester, as compared with their control mean magnesium value of 1.66 mEq/liter ± S.D = 0.01. They expressed concern about the magnesium deficiency early in pregnancy, at a time when hyperemesis gravidarum can lead to loss of minerals, including magnesium. They stressed the importance of repairing the magnesium deficit, as well as that of the fluids and more commonly considered electrolytes. This observation recalls the work of Hall (1957), who showed the lowest serum magnesium levels in the early weeks of his study, and that of DeJorge (l965b), who considered the magnesium deficit real only in the first half of pregnancy and the final month.
The change in serum magnesium that takes place during labor and in the parturient period are not clear. Wallach et al. (1962) found the concentrations of plasma Mg to be below normal in three normal parturient women (1.57-1.70 mEq/ liter), as compared with the normal value of 2.0 ± 0.15, obtained from 75 men and women 19-68 years of age. Celli Arcella (1965) reported that serum magnesium levels rose to normal levels during labor, after the low values they had noted during the third trimester. Lupi et al. (1967) noted low serum magnesium levels (1.4 mEq/liter) at the beginning of labor, but observed a further decline during the final stage of labor (1.1 mEq/liter). Manta et al.(1967) also found serum magnesium levels to decrease during labor, reaching the lowest point at the stage of expulsion and then rising. These findings confirm the early report that the mean serum magnesium levels drop at the end of labor to 1.5 [ range = 0.35-2.35 mEq/liter (Zaharescu Kamman, 1936b)], and those of Rusu et al. (1971/1973), who found that the mean serum magnesium levels dropped slightly at the outset of labor in 38 women to 2.0 mEq/liter from 2.3 just before labor began. During active labor there was a further drop (in 88 women) to 1.5 ± 0.3 mEq/liter. Ten women with imminent premature labor had a mean serum level of 1.4 ± 0.3 mEq/liter. The values depicted inTable 2-13 indicate that most investigators have found low maternal serum levels at delivery, cord blood values being significantly higher
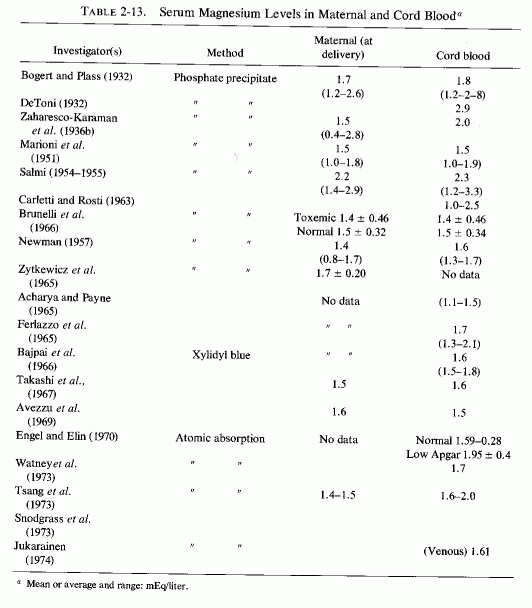{kind=link}
Caddell et al. (1973a) have evaluated the magnesium status of postpartum, well-fed women in Thailand (where the magnesium intake is greater than it is in the United States), and found that the postpartum plasma magnesium levels were significantly lower than they were in young nulliparous women. When they were tested by a parenteral magnesium load, the postpartum women retained a mean of 15% more magnesium than did the nulliparous women (borderline significance). Some apparently normal, asymptomatic postpartum patients had moderately high magnesium retention, but 37% retained only 0-25%. In a study of 198 moderate-income American mothers assessed by an intravenous magnesium load test, the mean postpartum magnesium retention was 51% (Caddell et al.1975). Over 90% of the magnesium load was retained by biologically immature (under 17 years of age) multiparas and in young mothers of twins. Most primiparous mothers showed little retention of the load, but 6 who had had prolonged labors retained 78% of the load. Multiparous mothers with a long interval since the previous pregnancy had the lowest magnesium retention. However, among the 46 patients who retained less than 40% of the load, the mean plasma magnesium was 1.58, and among the 81 who retained more than 40%, the plasma magnesium was 1.45 mEq/liter. Only plasma levels below 1.2 mEq/liter could be matched with high retention of magnesium.
The use of magnesium salts parenterally for control of manifestations of acute eclampsia long antedated the demonstration that serum levels of magnesium tend to be lower in women with toxemic pregnancies (especially early in the course of pregnancy) than they are during normal pregnancies. Less reliable as an index of magnesium deficiency of toxemic pregnancy is the serum level toward the end of gestation, when renal damage can interfere with magnesium excretion, as it does in patients with nephritis. The first published reports of the anticonvulsant properties of magnesium sulfate in eclampsia appeared in Europe (Einar, 1907; Kaas, 1917). It became a favored treatment of convulsions of pregnancy in the United States from the time Lazard (1925) and McNeile and Vruwink (1926) recommended its use intravenously, Dorsett (1926) described its use intramuscularly, and Alton and Lincoln (1925) reported its use intrathecally. Hirschfelder (1934) first reported a markedly low serum magnesium level (0.8 mEq/liter) in a 47-year-old patient with eclampsia, who then responded favorably to high dosage oral magnesium sulfate therapy. Among eight eclamptic women, Haury and Cantarow (1942) reported three with serum magnesium levels of 0.8-1.0 mEq/liter and three with levels of 2.7-3.2 mEq/liter. Their stages of pregnancy were not given. Achariet al. (1961) reported that 21 eclamptic women had a mean serum magnesium level of 0.83 mEq/liter (range = 0.25-1.84). Eclamptic women frequently have higher plasma or serum magnesium levels toward the end of pregnancy than do normal pregnant women at term (Pritchard, 1955; Hall, 1957; Kontopoulos et al. 1976/1979), but such normal or even elevated levels are not considered a contraindication to the use of large doses of magnesium salts, which are administered parenterally for their pharmacodynamic neurosedative, antihypertensive effects and not to correct a deficiency. As much as 200 mg of magnesium an hour, given intravenously as the sulfate, was recommended in the early studies (Lazard, 1925, 1933; McNeile, 1934; Winkler et al., 1942). This route is recommended by many either as the sole approach (Zuspan and Ward, 1964, 1965; Zuspan, 1966, 1969; Harbert et al., 1968; Hutchinson et al., 1963), or in combination with intramuscular injections (Pritchard, 1955; Flowers et al. 1962; Flowers, 1965, 1975; Kontopoulos et al., 1976/1980, Weaver, 1976/1980; Flowers et al., 1962, Fig. 2-3). Pritchard (1955) observed that administration of large doses of epsom salts orally exerted no effect on the plasma magnesium levels. Since only 5% of the administered dose appeared in the urine, the possibility that only a small percentage of the administered dose was absorbed was considered. However, even after administration of 150 g of magnesium sulfate intramuscularly over a five-day period, he found that the plasma levels were maintained between 3.5 and 7 mEq/liter. When treatment was initiated with 4 g of MgSO 4 intravenously, there was an initial peak, followed by a prompt rapid fall and then a gradual decline. He found that the cerebrospinal fluid magnesium levels did not reflect the high plasma levels induced by therapy. Flowers (1965) found it necessary to use a mean of 70 g of magnesium sulfate over a three-thy period to control eclampsia. Similarly, Harbert et al. (1968) found it necessary to use 40-60 g of magnesium sulfate per 24 hours to maintain neurosedative serum levels of magnesium of 6-8 mEq/liter. Perhaps the failure to develop hypermagnesemia more frequently toward the end of an eclamptic pregnancy and the difficulty in maintaining pharmacologic blood levels may reflect not only repletion of maternal stores but high fetal requirements, which might not have been supplied during the abnormal pregnancy.
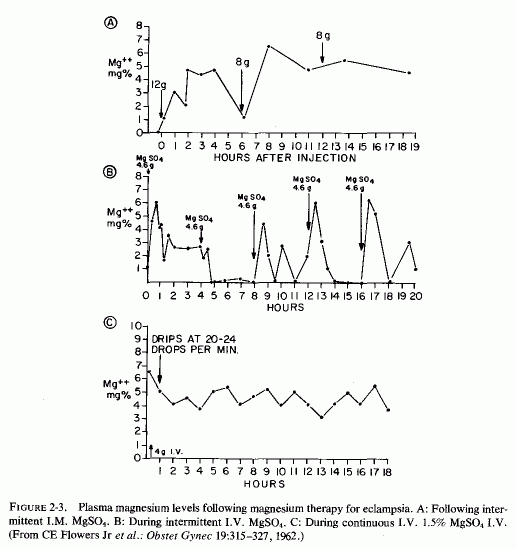{kind=link}
Hall (1957), because of the experimental and clinical evidence that magnesium deficiency is associated with neuromuscular irritability and convulsions, and because of the long-recognized efficacy of magnesium in the management of preeclampsia and eclampsia, considered the possibility that magnesium deficiency might contribute to toxemia of pregnancy. He found that the mean of plasma magnesium levels had been somewhat lower among toxemic than among normal pregnant women from the 12th through the 25th week. He charted a tendency of the magnesium levels to rise slightly toward the end of pregnancy in toxemic women (Fig. 2-2, Hall, 1957), a finding that might be related to increasing renal damage in that group. The percentage variations from the normal nonpregnant levels were as great as 50%-90% below the mean at different times during pregnancy. However, since the differences between the levels in the normal and toxemic pregnant women were not statistically significant, Hall questioned whether the low magnesium levels contributed to the symptoms of toxemia. Two years earlier a preeclamptic woman with pseudohypoparathyroidism (serum calcium of 4-6 mg percent and lack of response to PTH), and hypomagnesemia (1.1 mEq/liter) associated with mental aberrations, had been reported from the same medical center (Suter and Klingman, 1955). The possibility was considered that lowered serum magnesium levels during pregnancy might predispose to seizures during pregnancy in susceptible women, such as those with a tendency toward epilepsy (Suter and Klingman, 1957). Flowers et al. (1965) suggested that depletion of tissue stores of magnesium might explain eclamptic patients' tolerance and requirement for such large doses of magnesium. McGanity (1965) proposed that dietary magnesium deficiency might be etiologic in preeclampsia.
In France, where latent tetany had long been recognized as a manifestation of subacute magnesium deficiency (Durlach and LeBrun, 1959; 1960; Durlach, l969a) uterine cramps and abnormal contractility during pregnancy have been shown to be responsive to treatment with magnesium, and have been proposed as a manifestation of its deficiency (Dumont, 1965; Muller, 1968; Muller et al., 1971/l973).It was observed that patients with this complaint frequently also exhibited latent tetany and often had marginal hypomagnesemia (1.5 mEq/liter or lower levels), with and without hypocalcemia (Dumont, 1965). Uterine hypercontractility has been added to the signs of toxemia of pregnancy and has also responded to intravenous magnesium therapy (Hutchinson et al., 1963; Cobo, 1964).
The efficacy of pharmacologic doses of magnesium in the treatment of manifestations of toxemias of pregnancy has led to consideration of magnesium as a drug, in that condition, far more commonly than consideration of the fact that it is a nutrient, the supply of which must be increased substantially during gestation. Two years before hypomagnesemia was first reported in an eclamptic woman (Hirschfelder, 1934), magnesium deficiency was associated with abnormalities of pregnancy and during early lactation in cows (Sjollema, 1932). Neuromuscular manifestations in pregnant and lactating herbivores included tetany and convulsions; cardiovascular lesions were found at autopsy (Sjollema, 1932; Rook and Storry, 1962; Storry and Rook, 1962; Rook, 1963; Herd, 1966a,b; Hjerpe, 1971). Magnesium deficiency has been accepted as contributory to toxemia of pregnancy in grazing animals, and magnesium recognized as protective.
The possibility is increasingly being considered that magnesium deficiency can also contribute to major and lesser manifestations of toxemias of pregnancy (Dumont, 1965; McGanity, 1965; Lim et al., 1969b; Muller, 1968; Muller et al., 1971/1973; Hurley, 1971; Seelig, 1971; Seelig and Bunce, 1972; Kontopoulos et al., 1976/1980; Weaver, 1976/1980). There is evidence that the magnesium intake during pregnancy is likely to be suboptimal (Review: Seelig, 1971). That it might be sufficiently low to contribute to early and late abnormalities of pregnancy is suggested by the survey that showed magnesium intakes during pregnancy that are low (N. Johnson and Philipps, 1976/1980), even by standards for nonpregnant women (Seelig, 1964). The women with the lowest magnesium intakes gave birth to low-birth- weight infants, a finding that suggests intrauterine growth retardation. Mahran and Hanna (1968) expressed concern about the magnesium deficit, early in gestation, that might be caused by hyperemesis gravidarum. When one considers how frequently lesser degrees of nausea and vomiting (i.e., "morning sickness") interfere with proper nutrition in the first trimester, and one recalls the evidence that hypomagnesemia is encountered at that time (de Jorgeet al., 1965a,b) and shortly thereafter (Hall, 1957), the possibility of early magnesium deficiency being etiologic in abnormalities of pregnancy, placental abnormalities, intrauterine malnutrition, and fetal abnormalities should be seriously entertained. That hyperemesis can precipitate acute hypomagnesemia later in pregnancy was demonstrated in a report by R. Fraser and Plink (1951) of a 33-year-old woman who developed hypochloremic, hypokalemic alkalosis in association with hypomagnesemia a few days before delivery of her eighth child. It should be noted that so young a woman, completing her eighth pregnancy, would be expected to be magnesium depleted.
The abnormalities of placentas of eclamptic women which range from functional insufficiency (secondary to arteriolar spasm) to small size, scarring, and infarction (Warkany et al., 1961: Holman and Lipsitz, 1966; Wigglesworth, 1966) are associated with intrauterine growth retardation (IUGR) and hypoxia. There is insufficient evidence to implicate magnesium deficiency in eclamptic pregnancy as a direct contributory factor in placental abnormalities, but there are some findings that suggest the need for further study of this question. Charbon and Hoekstra (1962) tabulated the magnesium and calcium contents of placentas from women with normal single and twin pregnancies and with preeclampsia or eclampsia. The decreased magnesium and increased calcium levels of the placentas from eclamptic women are especially striking (Table 2-14). Magnesium-deficient pregnant rats had placental calcification and bore low-weight young (Cohlan et al., 1970; Dancis et al., 1971). An excess of vitamin D, which is known to cause net loss of magnesium (to be discussed later in this volume) has caused reduction in placental size in rats, placental damage, and birth of small for gestational-age young (Potvliege, 1962; Ornoy et al., 1968). Whether the peroxidized cod liver oil, or its fractions, that were used to produce experimental eclampsia in rats, with intravascular coagulation and damaged placental trophoblast (McKay et al., 1967) also cause magnesium loss has yet to be investigated. Changes similar to those caused by an excess of vitamin D or peroxidized vitamin D have been reported in placentas of women with eclampsia, and thrombocytopenia that reflects intravascular platelet aggregation of eclampsia has long been recognized (Review: McKay et al., 1967).
{kind=link}
The initiating factor that damages the syncytial trophoblast in human preeclampsia is not known. Hyperreactivity to vitamin D, possible formation of toxic derivatives of that sterol (Seelig and Mazlen, 1977), and the evidence that high Ca/Mg and Na/K ratios increase arterial resistance (Haddy and Seelig 1976/1980) are factors that should be considered. It remains to be resolved whether the observation that magnesium administration to preeclamptic women produced highly significant increased coagulation time and decreased platelet adhesiveness (Weaver, 1976/1980) indicates only a direct effect of magnesium on coagulopathy, or whether it plays a role in correcting a deficit that intensifies placental damage.
The coagulation abnormalities of eclampsia (McKay et al., 1967; Howie et al., 1976), the significant risk of antenatal thromboembolism (Editorial, Brit. Med., 1:249-250, 1970), and the hypercoagulability of the blood of eclamptic patients who have responded to magnesium therapy (Weaver, 1980) are provocative observations pointing toward a possible etiologic role of early magnesium deficiency. Perhaps the clinician is in error when he assumes that the hypomagnesemia seen during normal pregnancy is necessarily normal. It would have been useful had serum magnesium levels been obtained from the thriving mother whose sustained positive magnesium balance throughout pregnancy has been described by Hummel et al. (1936). Her daily intake of magnesium had exceeded the generally recommended 400 mg/day by 200 mg daily; she had retained 15½ g of magnesium during the last half of her pregnancy. If the magnesium requirements during pregnancy are as great as those suggested by the early Landsberg (1914) study and those of Hummelet al. (1936, 1937), and if magnesium deficiency contributes to toxemia, why is toxemia not more common? The answer may lie in the possibility that unlike the magnesium-deficient pregnant rats that sustain their own magnesium levels, with fetal loss, the normal less depleted woman draws more on her own reserves of magnesium to meet fetal demands. Her declining blood levels may reflect the drain, but need not be associated with maternal pathologic changes unless concomitant abnormalities are present. If the rat studies are relevant to pregnant women, it may be that in some women with insufficient magnesium deficiency to cause overt maternal changes, there can be fetal damage, in view of high fetal cellular demands for magnesium (McCance and Widdowson, 1961).
Already mentioned is the possibility that agents (such as vitamin D or derivatives) that affect magnesium requirements, as well as presenting other toxic potential (Seelig and Mazlen, 1977), can participate. Additionally, in a study of the effect of vitamin D with and without calcium and phosphorus supplements on success of gestation in rats, Nicholas and Kuhn (1932) found that unlike the test rats the control rats were given fresh green vegetables, yeast, fruits, butter, and cod liver oil and had the best gestations. Thus, the controls received a balanced diet containing magnesium, trace elements, the B vitamins, and vitamin A, which were absent in the experimental groups that had significantly less successful gestations. This study calls to mind the studies implicating pyridoxine deficiency in the "morning sickness" syndrome and in later manifestations of toxemia (Sprince et al., 1951; Klieger et al., 1966). The extent to which magnesium and pyridoxine deficiencies might interrelate in pregnancy-both nutrients are involved in phosphorylation reactions and protein synthesis (Review: Durlach, 1969b)-remains to be determined,
Even such a seemingly minor abnormality as a smaller than normal placenta has been associated with a disproportionate reduction in birth weight (Wigglesworth, 1966). Placental infarction, such as occurs in toxemic pregnancy, interferes with placental transfer of nutrients and affects gaseous diffusion, leading to lowered oxygen levels in the fetus. Scarred placentas have impaired blood flow, with resultant retardation of intrauterine growth and oxygenation (Walker and Turnbull, 1953; Warkany et al., 1961; Gruenwald, 1961, 1963, 1964; Scott and Usher, 1964; Holman and Lipsitz, 1966; Wigglesworth, 1966). Even moderate maternal malnutrition has been shown to be associated with significantly smaller than normal placentas and a high prevalence of low-birth-weight infants (Lechtig et al., 1975). Scott and Usher (1966) analyzed the factors associated with fetal malnutrition and found that it occurred in 13.5% of the primipara (usually young), and in only 8.4% of multiparous births, but that the incidence rose with each successive pregnancy after the sixth pregnancy. There was also a higher incidence of fetal malnutrition when previous pregnancies had produced low-birth-weight infants, to as high as a ninefold increased incidence when there had been four or more low-birth-weight infants. Infants with IUGR had a higher incidence of fetal distress, asphyxia neonatorum, and congenital abnormalities than did normal-weight infants. Congenital anomalies were diagnosed in 17% of the 60 markedly underweight infants and in 31% of 35 who were markedly wasted. The incidence represents a 30-fold increase in major anomalies and a 16-fold increase in congenital heart disease in infants with marked fetal malnutrition.
Placental insufficiency, found not only in eclampsia and frequent pregnancies but in prolonged gestation, placenta praevia, and pregnancy in the elderly primigravida patient, is associated with fetal malnutrition and low calcium and glucose levels in the infant (Khattab and Forfar, 1971). There should be routine determinations of magnesium levels and retention during and after pregnancy in women at risk of placental pathology, and of their infants.
Women who have experienced one spontaneous abortion not infrequently experience repeated abortions. Zigliara et al. (1971/1973) studied the magnesium levels of 294 women with imminent abortions and found that 50% had significantly lower than normal erythrocyte magnesium levels; 25% had hypomagnesemia as measured by serum determinations. The serum levels showed the existence of severe chronic magnesium deficiency in only 11% whereas low erythrocyte magnesium levels were seen in 40.8% of those with repeated abortions. The greater the number of abortions, the greater the degree of magnesium deficiency detected. Normal levels were reached three days after the abortion. Rusu et al. (1971/1973) also reported lower (than in normal pregnancy) magnesium levels among women with imminent abortions (1.4 ± 0.3 mEq/liter versus 2.0 mEq/liter). Treatment with magnesium doubled the serum levels and permitted some of the women to continue to term.
Whether the uterine hypercontractility, considered part of the preeclamptic syndrome (Hutchinson et al., 1963; Cobo, 1964) and found as a complication of pregnancy among women with latent tetany of marginal hypomagnesemia (supra vide), is related to the hypomagnesemia of recurrent aborters remains to be proved. Rusu et al. (1971-1973) found that as the serum magnesium level fell the uterine reactivity to oxytocin increased.
It may be relevant that magnesium-deficient animals have poor gestational success, with evidence of resorption at implantation sites in severely deficient animals, and smaller-than-control size of litters in less deficient animals.
The severity of postpartum uterine cramps has also been related to the drop in serum magnesium after delivery. Nicolas (1971/1973) reported that there was a slight decrease of magnesium levels during labor [1.74 (1.4-2.2 mEq/liter)] and 24 hours later [1.64 (1.4-2.2)]. A double-blind study in which one group was given magnesium therapy after delivery (500 mg magnesium lactate four times a day) or placebo resulted in a significant (p < 0.001) increase of magnesemia (from 1.6 to 1.9) in the magnesium-treated group, a change that was associated with improvement in uterine discomfort; there was no change in uterine cramps in the placebo-treated group.
Part I: Chapter 3
MAGNESIUM DEFICIENCY DURING GESTATION, INFANCY, AND EARLY CHILDHOOD
In view of the evidence that inadequate magnesium intake is common during pregnancy and that the plasma levels of magnesium tend to fall, especially during the first and third trimesters of pregnancy even when corrected for hemodilution, it is not surprising that neonatal magnesium deficiency can create problems. Until relatively recent years, however, measurement of magnesium levels in infants was rare. Cord blood analyses, done at intervals since 1923 (Table 3-1) and (Table 3-1 continued)showed wide ranges reported in individual studies, even when the quite reliable old precipitation methods or the more reliable modem procedures were employed. Since individual maternal status and infant status were not designated in most instances, these wide ranges are difficult to interpret. Low levels may have reflected maternal and fetal insufficiency; high levels may have reflected magnesium therapy for preeclampsia. Mean values are even more difficult to evaluate. Determination of serum or plasma magnesium levels of the infant at birth or within hours thereafter presents more problems. Intrauterine asphyxia, difficulties in delivery, or other causes of birth hypoxia or acidosis, and hyperosmolality can all contribute to elevations of serum magnesium levels as the cellular magnesium is released to the extracellular fluid, changes similar to those seen with surgical and other traumatic shock and hypoxic conditions. Such infants have been found to have a negative correlation between their serum magnesium levels and their Apgar scores (Engel and Elm, 1970; Jukarainen, 1974). Infants who are hypermagnesemic when born shortly after their eclamptic mothers had received pharmacologic parenteral doses of magnesium also are likely to be depressed and have low Apgar scores. The first group of infants is likely to be cellularly depleted of magnesium, which becomes manifest as hypomagnesemia, usually by the fifth day of life. Those with hypermagnesemia following maternal magnesium therapy usually take longer for their serum levels to drop to normal limits. If the infant survives the respiratory depression of pharmacologic hypermagnesemia, it is moot whether the presumed antenatal magnesium deficiency might have been corrected.
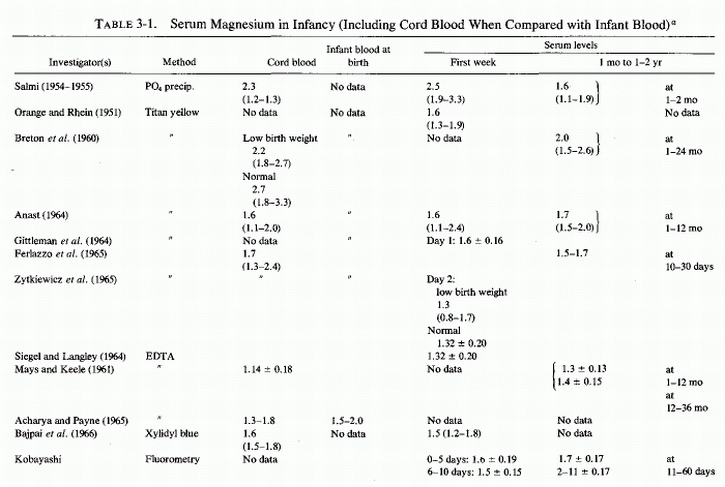{kind=link}
{kind=link}
Magnesium determinations during infancy have not been frequently reported; when reported, they have rarely included data on the maternal or infant status (Table 3-1) and (Table 3-1 continued). The first report found was one in which 24 magnesium levels were included in a table of 116 infants and children with a variety of abnormalities whose calcium levels had been analyzed (Denis and Talbot, 1921; Table 3-2). When the syndrome of magnesium malabsorption was recognized and infantile hypocalcemia was found often to be unresponsive to calcium or calcemic agents but to respond to magnesium repletion, magnesium determinations were done more commonly. The change in infant feeding patterns from breast-feeding to use of a variety of formulas has led to increased mineral retentions that are not paralleled by calcium and magnesium plasma levels, which are lower in infants fed cows' milk than in normal infants who are breast-fed.
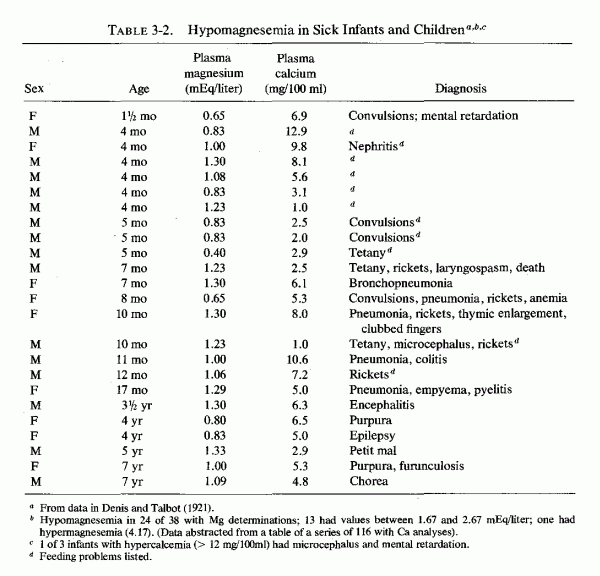{kind=link}
Infants at greatest risk of neonatal hypomagnesemia are low-birth-weight infants, including those suffering from intrauterine growth retardation (IUGR) or premature infants recovering from birth hypoxia or later respiratory distress, and infants born to very young primiparous women or to young mothers who have had frequent pregnancies or multiple births, to preeclamptic mothers, and to diabetic mothers. Plasma magnesium levels are a less reliable index of magnesium deficiency than is the parenteral load test, and magnesium deficiency, so demonstrated, has been found to be more common in newly born premature than in full-term infants, even when not indicated by notable hypomagnesemia (Caddell, 1975). The incidence of neonatal magnesium insufficiency may be greater than suspected. The tendency of women with preeclampsia or eclampsia to develop rising plasma magnesium levels during the last month of pregnancy, even without magnesium therapy, despite which they retain high percentages of parenterally administered pharmacologic doses of magnesium, suggests that magnesium deficiency might be far more common during pregnancy than is indicated by the incidence of hypomagnesemia.
To attribute the high incidences of placental and fetal abnormalities, stillbirths, and neonatal deaths (found among infants born to eclamptic women) to magnesium deficiency during gestation would be highly speculative at this stage of our knowledge. However, there are provocative findings that point to the possibility that it is likely to be contributory, not only to complications of pregnancy, but to damage to the products of conception. Interrelationships with other factors must be considered.
Rats kept severely magnesium depleted (receiving 1/200 the control magnesium intake) for the entire 21-day period of gestation had no living fetuses at term (Hurley and Cosens, 1970, 1971; Hurley, 1971; Hurley et al., 1976). The shorter the duration of the magnesium deficiency, the fewer implantation sites were affected. When the deficiency was maintained from day 6-12, about 30% of the implantation sites were involved and 14% of the full-term fetuses had gross congenital abnormalities (cleft lip, hydrocephalus, micrognathia or agnathia, clubbed feet, adactyly, syndactyly, or polydactyly, diaphragmatic hernia, and heart, lung, and urogenital anomalies). Milder magnesium deficiency (1/130 control intake) maintained throughout pregnancy resulted in resorption of half the implantation sites and malformation of the living young at term. In addition to their congenital anomalies, the surviving young were anemic and edematous. Surprisingly, despite the marked fetal damage caused by the deficiency during gestation, the pregnant rats showed only mild signs of magnesium deficiency despite sharp drops in their plasma magnesium levels. The severity of fetal damage produced in these studies was greater than in other studies; there might have been concomitant trace element deficiencies. Magnesium deficiency (1/130 of control intake), comparable to that produced in the less severely depleted rats of Hurley et al. (1976), but produced by adding salt mixtures with only the magnesium contents differing, resulted in less severe damage (Dancis et al., 1971; Cohlan et al., 1970). When the magnesium-deficient diet was fed from the second day of gestation to term, only one of eight rats bore a litter; the remainder had evidence of resorption at implantation sites. When the magnesium-deficient diet was fed from ninth or tenth day to term, the magnesium-deficient rats all produced live litters (8.1/litter), but there were also 36 resorption sites among the 17 test rats. The control rats had no resorption sites and delivered 8.5 pups/litter. The pups born to deficient dams were small (2.6 ± 0.1 g, in comparison to control mean weight of 3.8 ± 0.3 g) and were weak and pale. There was consistent microcytic anemia; edema was prominent in the severely anemic fetuses. The control fetuses had higher plasma magnesium levels than did the fetuses of the magnesium-deficient rats, a finding suggesting that there is relatively little protection of the fetus against maternal magnesium deprivation. The mothers looked healthy at term, and although they had hypomagnesemia, their tissue magnesium levels were only slightly lowered. In contrast, the fetal tissues were markedly magnesium depleted (Table 3-3). There was little difference in placental magnesium in the control and deficient groups, but the placental calcium of the magnesium-deficient fetuses also had higher tissue calcium levels (105 as compared with 80.1 in control fetuses).
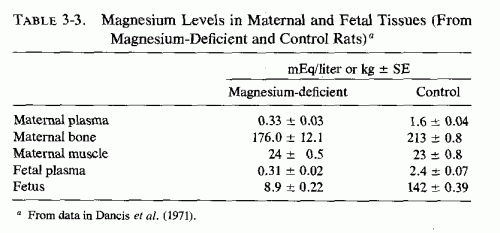{kind=link}
The less severe magnesium-depletion gestation study of Wang et al. (1971), which provided 1/10 the control amount of magnesium to deficient rats, did not significantly reduce the number of offspring but markedly reduced their viability. Labor was prolonged in the depleted group, and 53 (36%) of the 146 offspring were stillborn. By the fifth day after birth, 82 more had died spontaneously or been eaten by their mothers; only 7.5% survived. There were no obvious abnormalities, other than small size and occasional swelling of extremities. The deficient mothers were normal in weight but had significantly lower-than-control levels of serum and bone magnesium. They also exhibited impaired lactation, and secreted milk significantly lower in magnesium than that of control rats. The survival of pups fed by the magnesium-deficient dams was poor.
Dancis et al. (1971) speculated that the higher placental and fetal calcium levels of the magnesium-deficient rats might have reflected increased fetal parathyroid activity.
There is mounting evidence of magnesium insufficiency during pregnancy. Experimental acute magnesium deficiency has caused increased parathyroid secretion and even parathyroid hyperplasia (Larvor et al., 1964a; Kukolj et al., 1965; Gitelman et al., 1965, 1968a,b; Lifshitz et al., 1967; Sherwood et al., 1970, 1972; Targovnik et al., 1971). Thus, the possibility that magnesium deficiency is contributory to hyperparathyroidism of pregnancy, which is common despite widespread supplementation with calcium and vitamin D at that time, should be considered.
Low normal or subnormal plasma phosphorus levels during pregnancy, which rise postpartum, have long been associated with maternal hyperparathyroidism (Mull and Bill, 1934, 1936; Mull 1936; Bodansky, 1939). This condition has been found so frequently as to be termed "physiologic" (Hamilton et al., 1936; Cushard et al., 1972). Rat studies have shown that pregnancy can cause significantly increased parathyroid volume (Opper and Thule, 1943). Significant maternal hyperparathyroidism has been demonstrated by immunoreactive parathyroid hormone (PTH) measurements, the levels of PTH being significantly higher during the third trimester and at delivery than in age-matched nonpregnant women and than in cord blood (Samaan et al., 1973). Samaan et al.(1973) suggest that maternal hyperparathyroidism might be a response to the high fetal needs for calcium during the third trimester.
Despite hyperparathyroidism, serum calcium and magnesium levels both tend to be subnormal, especially during the third trimester of pregnancy (Watchorn and McCance, 1932; Mull and Bill, 1934; Mull, 1936; Bodansky, 1936; Kerr et al., 1962; Newman, 1957; DeJorge, 1956b; Lim et al., 1969b), which suggests that the gestational hyperparathyroidism can be secondary to hypocalcemia and/or to hypomagnesemia rather than physiological. Hyperparathyroidism has been found in mothers of infants with neonatal hypomagnesemia and hypocalcemia (J. A. Davis et al., 1965; Ertelet al., 1969; Monteleone et al., 1975).
Ludwig (1962) reviewed the relationship of hyperparathyroidism to gestation and the products of conception, and found that there was a greatly increased incidence of complications of pregnancy and of fetal loss and infant morbidity among diagnosed hyperparathyroid women. Since asymptomatic hyperparathyroidism (with gestation-hypocalcemia) is common during even normal pregnancy and has been implicated in symptomatic infantile hypocalcemia, the possibility should be considered that there might be a common denominator that contributes to both. The importance of calcitonin secretion, both in the mother and the neonate, is gaining increasing recognition. The role of magnesium deficiency during gestation should also be considered, since it is involved in parathyroid dysfunction and in calcitonin secretion. Maternal abnormalities that predispose to neonatal convulsive hypomagnesemic hypocalcemia include maternal magnesium deficiency, which predisposes to maternal hyperparathyroidism and is apt to occur in: (1) adolescent or young mothers (whose own magnesium requirements may not be fully met); (2) preeclamptic or eclamptic women; (3) women who have had several pregnancies in rapid succession, or with multiple births; (4) mothers with malabsorption; and (5) women with diabetes mellitus (Reviews: Tsang and Oh, 1970a; Tsang, 1972; Tsang and Steichen, 1975; Tsang et al., 1977a,b). Intrinsic (pregestational) hyperparathyroidism, of course, falls into this category.
The possibility that magnesium deficiency of pregnancy might be contributory to both transitory and sustained maternal hyperparathyroidism (with low serum calcium levels) should be considered, and the response to magnesium administration investigated.
The fact that cord blood phosphate, magnesium, and calcium levels are usually higher than maternal levels (Bakwin and Bakwin, 1932; Finola et al., 1937; Bruck and Weintraub, 1955; Delivoria-Papadopoulos, 1967; Samaan et al., 1973; Bergman, 1974; David and Anast, 1974; Tsang et al., 1973b, 1976b) suggests that fetal homeostasis of these elements is at least partially independent of maternal factors. Maternal hyperparathyroidism has long been speculated to be a direct or indirect cause of neonatal hypoparathyroidism, which contributes to hyperphosphatemia and secondary hypocalcemia and hypomagnesemia that are seen in the early days to weeks of life (Friderichsen, 1938, 1939; Van Arsdel, 1955; Hutchin and Kessner, 1964; Hartenstein and Gardner, 1966; Mizrahiet al., 1968; Ertel et al., 1969; Tsang et al., 1973a). Severe enough experimental hyperparathyroidism in pregnant rats, however, to cause hypercalcemia and renal and myocardial damage in the mothers caused no more fetal hypercalcemia than was seen in control fetuses and caused no fetal soft tissue calcinosis (Krukowski and Lehr, 1961a,b; Lehr and Krukowski, 1961), suggesting that the placental barrier protected the fetus against maternal hyperparathyroidism. These investigators reviewed the literature at that time, and discussed the early experimental evidence that PTH does not penetrate the placental barrier, either from the maternal to the fetal circulation, or from the fetus to the mother. Earlier, Hoskins and Snyder (1933) showed that injection of PTH into the dog fetus in utero resulted in elevated fetal serum calcium levels not associated with a simultaneous rise in maternal plasma calcium levels. PTH injection into the pregnant dog raised maternal but not fetal calcium levels. An accidental finding during another study of the effect of hyperparathyroidism in dogs was obtained when one of the dogs was found to be pregnant (Cantarow et al., 1938). The absence of damage to the fetuses, such as had been produced by PTH in the mother, was interpreted as indicating possible lack of passage of PTH through the placental barrier. When they confirmed these findings in their own controlled experiments with rats (Lehr and Krukowski, 1961; Krukowski and Lehr, 1963), they judged that since the placental membrane is at least three cell layers thick (Wislocki and Dempsey, 1955) even at the time of maximal placental permeability, large proteins such as PTH are unlikely to penetrate it. This hypothesis has been proved correct. Garel and Dumont (1972) have shown no demonstrable maternal-fetal or fetal-maternal crossover of tagged PTH in the rat. Injection of PTH to fetal rats has raised their serum calcium levels, and influenced their serum magnesium and phosphate levels (Garel, 1971b; Garel and Barlet, 1974). The effect of PTH injections into the rat fetus suggests that it mobilizes bone calcium, as indicated by exposure of fetal rat bones to PTH (Raisz and Niemann, 1967, 1969). Garel and Barlet (1974) were unable to confirm earlier observations that PTH decreases fetal plasma phosphate (Garel and Geloso-Mayer, 1971), and speculated that mobilization of bone mineral by FTH should increase fetal plasma phosphate levels.
In contrast to the inability of maternal PTH to cross the placenta, calcium and magnesium are readily transferred across the placental barrier (MacDonald et al., 1965). Their higher levels in fetal than in maternal blood suggest that there is active placental transport from maternal to fetal circulation (Economu-Mavrou and McCance, 1958; Aikawa and Burns, 1960; Cohlan et al., 1970). An active placental transport mechanism involving calcium and magnesium-stimulated ATPase has been identified (Whitsett et al., 1977a,b).
Inferential evidence has been obtained that modulation of increased or decreased fetal parathyroid activity protects the fetus against maternal hyper- or hypocalcemia and hyper- and hypophosphatemia, whether induced by dietary means, by maternal parathyroidectomy (Sinclair, 1942), high doses of PTH (Lehr and Krukowski, 1961; Krukowski and Lehr, 1963), or by hypervitaminosis D (Potvilege, 1962). The same should be true for protection against hyper- or hypomagnesemia. More recently there has been experimental proof that fetal parathyroids are functional. Garel and Geloso-Meyer (1971) demonstrated that thyro- or parathyroidectomy of pregnant rats causes fetal as well as maternal hypocalcemia, secondary fetal parathyroid hyperplasia, and resultant rises in the fetal plasma calcium levels. Ablation of the fetal parathyroids or injection of anti-PTH serum into the rat fetus (Garel, 1971a) has resulted in sustained fetal hypocalcemia. Production of fetal hypocalcemia by injection of the disodium salt of EDTA (which also chelates magnesium, although this was not measured) into sheep fetuses caused increased fetal PTH levels, but no change in the maternal PTH levels. In contrast, infusion of EDTA to normocalcemic ewes in late pregnancy caused a marked reduction in maternal plasma unchelated calcium and a doubling of maternal PTH levels, but no significant change in either of these parameters in the fetuses. Infusion of calcium to the pregnant ewes lowered their PTH levels but caused no change either in calcium or PTH levels of their fetuses (Care et al., 1975). Studies in monkeys, however, have shown that fetal serum PTH was undetectable in the basal state and in response to EDTA-induced fetal hypocalcemia, although EDTA-induced maternal hypocalcemia caused 30-197% increases in maternal PTH values (A. R. Fleischman et al., 1975). Whether the presumed simultaneously reduced serum magnesium levels interfered with release of PTH from the fetal glands requires investigation. Garel and Barlet (1976) have shown species differences in the parathyroid status at birth. Thus, there should be caution in applying experimental findings to human perinatal hormone/mineral interrelationships.
Much less work has been done on the fetal parathyroid response to low magnesium levels. Since fetuses of magnesium-deficient rodents show more damage than do the mothers, it seems likely that fetal parathyroid activity is less effective in protecting the fetus against hypomagnesemia than against hypocalcemia.
Hyperparathyroidism of pregnancy has long been blamed for hypoparathyroidism and low serum calcium/phosphorus ratios in the neonatal period (Friderichsen, 1939; Bakwin, 1939). The existence of neonatal tetany is considered a sensitive clue to maternal hyperparathyroidism. Hartenstein and Gardner (1966) reviewed the literature and found that there were seven reported families, including their own reports, in which neonatal tetany was associated with maternal parathyroid adenoma. Friderichsen (1939) was the first to report the association in an infant who developed infantile tetany at five months of age, and whose mother had osteitis fibrosa cystica secondary to her parathyroid adenoma. Brief reference was made to unusually severe signs of hypocalcemic neonatal tetany on the second day of life of two infants born to hyperparathyroid mothers (Talbot et al., 1954). Maternal symptoms can well be absent in hyperparathyroid mothers whose premature or full-term infants present with severe tetany (Walton, 1954; Van Arsdel, 1955). A mother of eight children (four of whom had had hypocalcemic neonatal tetany developing at the 14th, 12th, 9th, and 2nd days) who had another pregnancy that aborted had asymptomatic parathyroid adenoma that was not diagnosed until her renal calcinosis was found a year and a half after the birth of her last child (Hutchin and Kessner, 1964). Conversely, infantile hypocalcemic tetany did not develop until one year of age in an infant, three months after cows' milk was substituted for breast milk, which had been provided by his mother who had had symptoms and signs of hyperparathyroidism (Bruce and Strong, 1955). Hypoparathyroidism was diagnosed in that child in his fourth year of life; a parathyroid adenoma was removed from the mother six years after he was born.
It was first suggested by Pincus and Gittleman (1925) that transient hypoparathyroidism might be at fault in a seven-week-old infant with nonrachitic tetany. Bakwin (1937) considered the susceptibility of neonatal infants to hyperphosphatemia and secondary hypocalcemic tetany to be a result of the phosphate load provided by cows' milk fed to infants with end-organ unresponsiveness to PTH at birth. However, fetal plasma phosphorus levels are usually considerably higher than are maternal plasma levels (McCance and Widdowson, 1954, 1961), and even breast-fed infants who do not have a free supply of milk during the first 48 hours show a rise in serum inorganic phosphate after the first 24 hours (McCance and Widdowson, 1961). This rise has been attributed to the expenditure of tissue glycogen and protein to maintain life while the intake is minimal, an observation that has been supported by study of fasting newborn pigs (McCance and Widdowson, 1957). Before and at birth (cord blood) there are elevated fetal or infant plasma phosphorus levels that are associated with higher than maternal plasma levels of calcium and magnesium (Reviews: Smith, 1959; Bergman, 1974; Tsang et al., l976b).
3.2.3.1. Hypocalcemia of Infancy
A few hours after birth, infants commonly exhibit sharp drops in plasma calcium levels (Review: L. Bergman, 1974). Their phosphate levels tend to remain high for days to weeks, especially those fed cows' milk. This is seen in normal full-term infants but is particularly marked in low-birth-weight infants, those that are born to diabetic mothers, or those that have been born after difficult deliveries and suffered birth hypoxia or later respiratory distress. It has been stressed that early neonatal hypocalcemia should be distinguished from that developing only after a week of life or later, which is related to the phosphate load of cows' milk. The foregoing section on neonatal and persistent infantile hypoparathyroidism [particularly with reference to the four siblings who developed the syndrome at 2-14 days (Hutchin and Kessner, 1964)] suggests that there might be a common denominator for both, and that the phosphate load precipitates the syndrome in less abnormal infants.
The greater predilection for hypocalcemia and hyperphosphatemia among premature than full-term infants, and the rarity with which breast-fed infants develop these abnormalities within the first three weeks of life, were clearly depicted by Bruck and Weintraub (1955). Both groups had lower calcium levels after birth than they had had in their cord blood. Few of the premature hypocalcemic infants had tetanic symptoms; they more commonly presented with convulsions, hypersensitivity, rigidity, edema, vomiting, respiratory disturbances, and drowsiness. However, there were frequently no abnormal symptoms. The authors cautioned against considering asymptomatic hypocalcemia as "physiologic," since sudden transition from latent to manifest tetany is frequent. In the 1918 review of infantile tetany by Howland and Marriott, they reported four publications on the syndrome from 1815 through 1887. They were the first to observe that the syndrome could occur in the absence of rickets, and that it was far more common in cows'-milk-fed than in breast-fed infants. Dodd and Rapaport (1949), in their review, reported only sporadic cases from 1913. Among their own series of 33 infants with symptomatic neonatal hypocalcemia, 22 had convulsions, 28 had vomiting, 16 had edema (severe in 9), and 12 were cyanotic. Hemorrhagic manifestations included hematemesis (6 cases), melena (4), and hemoptysis or petechiae (2). Saville and Kretchmer (1960) commented on the rarity of reports of neonatal tetany until late in the 1930s, and its increasing frequency thereafter. They reviewed the evidence that a combination of cows' milk and vitamin D supplementation, together, were potent means of inducing infantile hypocalcemia and considered the high incidence in the literature among infants born after difficult labor or to diabetic mothers. They confirmed these observations in their series of 125 cases in a major medical center from 1940 to 1958. Only 33% were the products of normal full-term pregnancies and uneventful labor. Almost a tenth were born to diabetic mothers. Both low-birth-weight infants and those born to diabetic mothers, as well as other "sick" and hypocalcemic infants, have been shown to have subnormal parathyroid function (L. David and Anast, 1974; Samaan et al., 1973; Tsang et al., 1973b, 1975a, 1976a, 1977a; Bergman, 1974; Bergman et al., 1974; David et al., 1976, 1977).
It has been speculated that the hypoparathyroidism of infancy might be related to parathyroid immaturity (especially in premature or dysmature infants), to functional parathyroid deficiency, or to fetal hypercalcemia, possibly deriving from maternal hyperparathyroidism-induced hypercalcemia that might cause fetal PTH suppression, mediated by resultant fetal hypercalcemia (Reviews: Tsang et al., 1973a, 1976a).
On the basis of the early experimental evidence as to fetal parathyroid competence, Lehr and Krukowski (1961b, 1963) commented that it is invalid to blame the neonatal rise in serum phosphate, with resultant drop in serum calcium, on the inability of functionally immature neonatal parathyroids to compensate for the loss of maternal PTH. They suggested that the difference between maternal and fetal serum calcium levels might reflect the higher pCO2 in fetal blood, a deduction made on the basis of their observation that hypoxic fetuses (taken from dams after sacrifice) had markedly higher serum calcium levels than did fetuses without hypoxia (taken from living anesthetized dams) (Krukowski and Lehr, 1963). They proposed that the drop in serum calcium to normocalcemic levels immediately after birth might be mediated by initiation of respiration with blowing off of excessive CO2 It is of interest, in this regard, that correction of neonatal acidosis by administration of bicarbonate in premature infants (Tsang and Oh, 1970a; Tsang et al., 1976b), in infants with intrauterine growth retardation who often have asphyxia (Tsang et al., 1975a), and in infants (often with respiratory distress) of diabetic mothers (Tsang et al., 1974) results in further reduction in serum calcium, with production of continued hypoparathyroidism. These findings call to mind the hypothesis of Barzel (1971) that PTH function is influenced by the bicarbonate/carbonic acid buffer system. He has shown that hypoparathyroid patients have simultaneously elevated plasma phosphate and pCO2 levels, with normal blood pH. However, the persistence of hypoparathyroidism, despite both hyperphosphatemia and hypocalcemia in infants whose elevated pCO2 and acidosis have subsided, suggests that another mechanism can be operative. It is unlikely to be neonatal parathyroid immaturity; fetal parathyroid function has been shown to protect the fetus against experimental maternal aberrations in phosphate, calcium, and magnesium levels; and immunoreactive evidence of fetal PTH has also been obtained (supra vide).
Nonetheless, low plasma PTH levels have been demonstrated in the first day or two of life (Tsang et al., 1973b; Samaan et al., 1973; L. David and Anast, 1974; Root et al., 1974; Tsang et al., 1975a), indicating failure of PTH secretion or release during the early neonatal period. Tsang et al. (1973b) have shown that PTH levels did not increase during the 24- to 48-hour period during which serum calcium levels fell. They found no relationship between serum PTH levels and total and ionized calcium in maternal, cord, and infant sera. Less gestationally mature infants had less increase in serum PTH during hypocalcemia than did the more mature infants. In contrast, David et al. (1976, 1977) showed that low-birth-weight infants had higher immunoreactive (i) PTH levels at birth than did normal adults, and that the iPTH levels increased earlier and were higher than in normal full-term infants (David and Anast, 1974; David et al., 1977). These investigators commented on the difference between their findings and those of Tsang et al. (1973), whose study infants were more severely hypocalcemic than were those of David et al. (1977). Additionally, the infants in the French study (David et al., 1977) were breast-fed; those in the American study (Tsang et al., 1973b) were bottle-fed. Possibly the absence of hyperphosphatemia and hypomagnesemia in the French infants might reflect the difference in the feeding customs. It is conceivable that the rise in PTH in premature rhesus monkeys as early as six hours after delivery (Fleischman et al., 1975) might be similarly explained.
The evidence that magnesium deficiency during gestation and in the neonatal period can be correlated with parathyroid dysfunction suggests that magnesium deficiency might well be an important contributory factor to infantile hypoparathyroidism, failure of target organ response to PITH, and to hypocalcemia. Inadequate supply of magnesium to the fetus can result from insufficient maternal intake, abnormalities of pregnancy during which there is subnormal maternal magnesium or placental damage that interferes with transport of nutrients including magnesium to the fetus. High-risk infants, usually born to mothers with abnormalities of pregnancy, have a high incidence of transient or prolonged hypoparathyroidism with symptomatic neonatal hypocalcemia. Their magnesium deficiency is usually detected later, either as hypomagnesemia, often after calcemic agents have failed to control neuromuscular irritability, or by demonstration of high percentage retention of parenteral loads of magnesium.
Hypoparathyroidism was reported in two infant sisters (children of first cousins), in association with severe hypomagnesemia (0.5 and 0.4 mEq/liter, respectively) that was detected subsequent to treatment of their hypocalcemia with high doses of vitamin D (100,000 U/day) or dihydrotachysterol (Niklasson, 1970). Despite the calcemic agents, their serum calcium levels rarely reached normal or hypercalcemic levels. One exhibited mental retardation and emotional lability at 20 months of age. Convulsions were common in this family, a finding that suggests that there may have been a genetic abnormality in magnesium metabolism. The correlation of maternal magnesium deficit with maternal hyperparathyroidism, and with neonatal hypoparathyroidism and hypomagnesemic hypocalcemic tetany and convulsions, is inferential evidence that the infants reported by Niklasson (1970) are not likely to be unique. David and Anast (1974) showed immunoreactive PTH levels to be low during the first nine days of life in normal, "sick," and hypocalcemic infants. They found that depressed plasma magnesium levels (range = 0.97- 1.25) were frequent (20%) in hypocalcemic infants. In normal newborn infants the range of plasma magnesium was 1.6-1.75 mEq/liter. These infants' hypomagnesemia was transient, usually reaching normal levels even when magnesium supplements were not given, or when the hypocalcemia was corrected by treatment with calcium. The rarer but more severe form of neonatal hypomagnesemic hypocalcemia associated with magnesium malabsorption must be treated with large magnesium supplements for correction of parathyroid suppression.
However, an infant has been described with the same syndrome but with hyperparathyroidism (Monteleone et al., 1975). The authors suggested that his seizures, which were intensified by calcium but responded to magnesium therapy, might have been causally related to hypomagnesemia secondary to his mother's hyperparathyroidism. They regretted that the PTH determination had been run after magnesium treatment had been started, thereby making it impossible to rule out the possibility of functional hypoparathyroidism immediately after birth. The infant's continued hypocalcemia and elevated PTH values suggested that he might have had target organ unresponsiveness to PTH, such as has been reported in magnesium-depleted older patients. They referred to the suggestion of L. Chase et al. (1974) that hypomagnesemic patients with hypocalcemia might have impaired skeletal response to PTH, with decreased heteroionic exchange of magnesium at the bone surface, a hypothesis proposed also by Zimmet (1968), who cited Neuman and Neuman (1957) regarding the theory that cation exchange for calcium occurs predominantly at the hydration shell.
3.3.1. Calcitonin during Pregnancy
Calcitonin (CT) levels are higher in maternal blood at time of delivery than they are in nonpregnant women (Samaan et al., 1973a,b, 1975). Pregnant ewes have elevated CT levels during the last 40 days of gestation, both on a low- and high-calcium intake (Barlet, 1974; Barlet and Garel, 1974; Garel et al., 1974, 1976; Garel and Barlet, 1975). Since the highest levels have been found in the ewes bearing triplets, there is support for the suggestion (Lewis et al., 1971) that CT might function to protect the bones of pregnant or lactating females against excessive demineralization (by the increased PTH of pregnancy) to meet fetal calcium needs. The response to hypercalcemia in pregnant animals is increased CT secretion. Infusion of calcium salts has augmented the CT secretion of pregnant ewes (Garel et al., 1973, 1974, 1976). Since pregnant women characteristically have hyperparathyroidism with hypocalcemia, as well as hypomagnesemia, the CT-stimulatory mechanism would appear not to be hypercalcemia. The hypocalcemia might reflect the response to CT secretion that spares the maternal skeleton. The mechanism by which CT secretion is increased in the presence of hypocalcemia, which (from the above studies) should decrease C-cell activity, remains unclear. Possibly, the simultaneously low magnesium levels during pregnancy play a role. For example, although magnesium-deficient rats show increased C-cell activity and release of CT in the presence of hypercalcemia (Stachura and Pearse, 1970), magnesium-deficient dogs with hypocalcemia also develop C-cell hyperplasia and evidence of increased secretory activity (Rojo-Ortega et al., 1971, 1971/1973). Thus, during human pregnancy, when plasma levels of both calcium and magnesium are low, conflicting responses might be responsible for both hyperparathyroidism and hypercalcitoninemia.
Maternal CT has been shown not to cross the placental barrier in rats (Garel et al., 1969, 1973, 1976) or in ewes (Garel et al., 1974). Fetal thyroid tissue is able to secrete CT, which exerts a hypocalcemic effect (Garel et al., 1968, Garel, 1969). That fetal C-cells can respond to hypercalcemia has been shown by Littledike et al. (1972) and Garel et al. (1973, 1974), who evoked significant increases in plasma CT in ovine, bovine, and porcine fetuses by acute elevations in fetal calcium levels, Administration of exogenous CT to the rat fetus, late in gestation, lowers the plasma levels of all three elements; calcium, magnesium, and phosphorus (Garel et al., 1968, 1969; Garel and Barlet, 1974). Samaan et al. (1975) attribute infantile hypocalcemia to elevated CT levels.
3.3.3. Neonatal Calcitonin
The level of immunoreactive CT (iCT) is significantly higher in the cord blood of full-term infants than in maternal blood at time of delivery following normal pregnancies (Samaan et al., 1973a,b, 1975). By use of a method of determination that does not detect iCT in normal children and adults (150 pg/ml), David et al. (1977) found just detectable levels in cord blood of low-birth-weight infants, with a marked increase after 1-2 hours of age, and a peak almost 5-fold higher by 11 hours after birth. Similar findings were reported in infants of diabetic mothers (Bergman, 1974; Bergman et al., 1974). Severalfold-higher plasma CT levels have also been detected in newborn lambs than in their mothers (Garel et al., 1974), and the levels have risen in response to calcium per os (Garel et al.1976) and in response to injection of cholecystokinin-pancreozymin (Barlet and Garel, 1976). Garel (1969) demonstrated that injection of CT into newborn rats produced a marked lowering of their plasma calcium levels. On the other hand, in subsequent work showing species differences in PTH/CT/Ca/Mg interrelationships in newborn ruminants, Garel and Barlet (1976) pointed out that the CT levels do not necessarily correlate with plasma calcium levels. Bergman (1974) postulates that high levels of growth hormone (i.e., in response to glucose infusion) at the time that CT levels are high increases the risk of neonatal hypocalcemia. Samaan et al. (1975) attribute infantile hypocalcemia to elevated CT levels.
The high blood levels of CT of neonates, and the preliminary evidence that CT secretion is increased in magnesium depletion (Stachura and Pearse, 1970; Rojo Ortega et al., 1971), as well as in the presence of high magnesium levels (Radde et al., 1970; Care et al., 1971; Littledike and Arnaud, 1971; S. P. Nielsen, 1971/1973, 1974; Barlet et al., 1974), suggests that early and sustained infantile hypocalcemia might be a function of combined hypomagnesemia/hypoparathyroidism/increased CT secretion-all of which respond to moderate doses of magnesium. The possibility that perinatal magnesium deficiency might be a contributory or even a fundamental abnormality in the mineral and hormonal aberrations of the perinatal period has received little consideration.
In the late 1930s, the decade that vitamin D supplementation became fairly commonplace, placental scarring and calcification were found to be more common in women supplemented with viosterol (vitamin D than in those who were not supplemented or who were given cod liver oil (vitamin D + vitamin A) (Brehm, 1937). The same year, Finola et al. showed that viosterol (vitamin D 250 U/day) given with calcium phosphate supplements caused little or no change in serum phosphorus levels as compared with the levels of those given the calcium salt alone, but several had serum calcium levels at or above 11 mg/100 ml (Fig. 3-1). Cord blood analyses showed a shift toward higher phosphorus levels and a higher incidence of hypercalcemia among infants of mothers given viosterol plus calcium phosphate than among those born to mothers on the calcium diphosphonate alone, although the averages were similar (Fig. 3-1). Both groups (Finola et al. 1937; Brehm, 1937) expressed concern about the tendency toward intrauterine osteosclerosis in the infants of the vitamin-D-supplemented mothers, which was associated with narrowed and closed fontanels, a finding they considered contributory to longer, more difficult labors. Of greater concern to Brehm (1937) was the placental calcification, scarcely notable in those who had not had vitamin D supplementation, but so marked among several of the women given viosterol plus calcium as to interfere with placental separation. Three stillborn infants with severe renal calcification were born to that group of mothers. No note was taken in either of these studies of the maternal intake or levels of magnesium, but studies of customary magnesium intakes at about that time suggest that intakes might not have been optimal. These are preliminary observations that should be tested, a difficult undertaking with American women because milk in the United States is almost universally fortified with vitamin D2 (400 units/quart) and calcium and vitamin supplements, providing 400 IU of vitamin D per tablet, are given to most pregnant women. This practice has been widespread since it was realized in the 1930s that failure to meet the demands for calcium, which increase manyfold during the last trimester, can cause maternal hypocalcemia (Cantarow et al., 1938). The need for prenatal vitamin D supplementation was predicated on the observation of rickets of the newborn (Coons and Blunt, 1930).
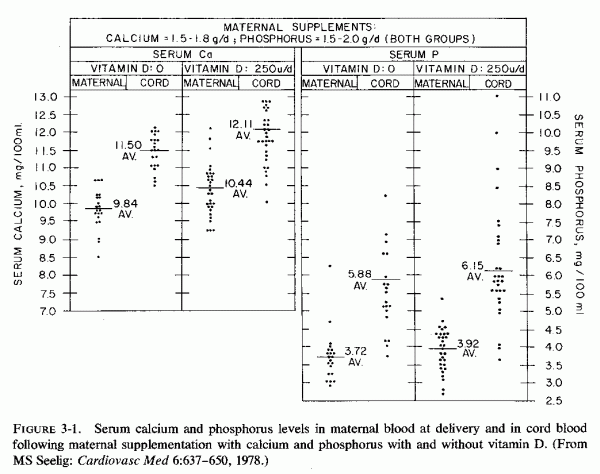{kind=link}
Subsequent work suggests that this practice might not be uniformly beneficial. Low magnesium intakes, such as are common during pregnancy, in combination with calcemic agents, favor a high Ca/Mg ratio. Experimental studies show that high Ca/Mg and Na/K ratios increase arterial resistance (Review: Haddy and Seelig, 1976/1980. Whether the high Ca/Mg ratio of intake during pregnancy might contribute to toxemic hypertension must be explored. Furthermore, normal and abnormal vitamin D metabolites have widely differing potency and toxicity (Seelig and Mazlen, 1977). Whether subjection of foodstuffs, to which vitamin D has been added, to a variety of cooking processes might convert the antirachitic factor to more toxic derivatives has not been investigated. It is known, for example, that peroxidized cod liver oil and some of its fractions can damage the placenta, with resultant intravascular coagulation and eclampsia in rats (McKay, 1962; Kaunitz et al., 1962, 1963; McKay and Goldenberg, 1963; McKay and Wong, 1962; McKay et al., 1967). Also, administration of high doses of even untreated vitamin D to rats has caused decreased placental volume, with atrophy and mucoprotein infiltration in the portion of the placenta composed of allantoic villi carrying fetal vessels (Potvliege, 1962). The fetal capillaries show degeneration of the endothelial cells, and the intervillous spaces collapse and become relatively bloodless. Calcium deposition occurs late in degenerated villi and in the walls and surrounding mesenchyma of large fetal vessels. Ornoy et al. (1968) has also shown that hypervitaminosis D in rats causes decreased placental volume. The young of the hypervitaminotic rats with placental pathology are small for gestational age, a finding similar to that seen in human infants born to eclamptic mothers or to others with abnormal placentas. Potvliege (1962) has found that there is also significant decrease in the volume of the parathyroids both in dams and fetuses, suggesting that the vitamin D might have caused hypercalcemia in both. The mothers had marked systemic calcinosis. The fetuses, however, showed neither vascular lesions nor excessive calcium deposition. In fact, pregnant rats that developed placental lesions (Ornoy et al., 1968) had fetuses with defective bone formation. These investigators attribute the anomalous bone formation to damage to fetal osteogenic tissues induced by passage of excessive vitamin D2 through the vitamin-impaired placenta. They speculate that vitamin D-impaired placental function permits excessive vitamin D to reach the fetuses, and presume that fetal damage is caused by vitamin D2 toxicity at the cellular level. W. Friedman and Roberts (1966) have shown that the blood levels of antirachitic sub stance are high both in rabbit mothers given toxic amounts of vitamin D2 and their young, but the fetal damage produced resembles more that seen in human babies during the epidemic of infantile hypercalcemia during a time of excessive vitamin D prophylaxis of rickets (Reviews: Black, 1964; Seelig, 1969b) than that seen in the rats. As with the rats, the does poisoned with vitamin D2 had greater damage than did their young, but the offspring had cardiovascular lesions: supravalvular aortic stenosis (Coleman, 1965; Friedman and Roberts, 1966; Friedman, 1968), endocardial thickening (Coleman, 1965), and premature closure of the fontanels, osteosclerosis, and palatal abnormalities (Friedman and Mills, 1969). It seems likely that both vitamin D3 and its 25-hydroxy-derivative cross the placental barrier from mother to fetus in the rat (Haddad et al., 1971). Judging from comparable 25-OH-D3 levels in maternal and cord blood, placental transport probably also takes place in humans (Hillman and Haddad, 1974; Belton et al., 1977). The observation that levels of 25-OH-D3 are lower in newborn rabbits with supravalvular aortic changes, born of does with hypervitaminosis D than they are in controls (Mehlhorn et al., 1977) suggests that administration of toxic amounts of vitamin D might result in its abnormal metabolism. It can be speculated that, as the enzymes involved in normal vitamin D metabolism are overloaded, abnormal degradation products can be produced. Whether there are such abnormal products, and whether they are more toxic than the normal metabolites should be investigated.
Unfortunately, although the enzyme systems involved in hepatic and renal hydroxylation of vitamin D are magnesium dependent, magnesium levels have not been determined in the studies of vitamin D toxicity in pregnancy, nor in the dam aged young. Since administration of high doses of magnesium is protective against vitamin D toxicity and magnesium deficiency intensifies the damage produced, the interrelationships of vitamin D and magnesium during pregnancy should be studied. Does magnesium deficiency increase the risk of vitamin D toxicity, and if so, to what extent? This is a cogent point, since the average American woman probably ingests considerably more than optimum quantities of vitamin D from fortified milk and other foods, as well as from prenatal supplements. Her intake of magnesium is likely to be marginal, at best, and is likely to be significantly low. Can magnesium supplements during pregnancy protect against vitamin D toxicity, and to what extent? This question might be relevant to protection against eclampsia, damaged placenta, and intrauterine growth retardation, as well as against fetal abnormalities-from bone to renal to cardiovascular anomalies-such as have been seen in experimental vitamin D toxicity during pregnancy, and some of which have been related to experimental magnesium deficiency itself. The nature of the fetal abnormalities caused by experimental hypervitaminosis D during gestation seems not be a function of the vitamin D alone, but to other components of the diet in ways that have not yet been clearly defined. The early study by Nicholas and Kuhn (1932) showed that their control pregnant rats given a complete diet that included fresh green vegetables and fruits, butter, yeast, and cod liver oil (a diet that was undoubtedly rich in magnesium, trace elements, and the B vitamins, as well as in vitamins A and D3 had uniformly successful gestations. To explore the influence of viosterol, calcium, and phosphorus, diets were prepared that lacked the above ingredients, and that provided adequate calcium and phosphorus and that were supplemented with or free of vitamin D2 (viosterol). Because of the absence of the additional nutrients in the "basic" experimental diet, the less successful gestations of the rats on that diet when viosterol was added reflects more than the influence of the vitamin D2 It is interesting, however, that the rats receiving the basic diet, adequate in calcium and phosphorus, did not tolerate the viosterol as well as did those on the diet that was deficient in calcium and phosphorus. When viosterol was given throughout pregnancy, none of the rats getting calcium and phosphorus delivered young; when given viosterol only during the last 14 days, two in ten rats came to term. One of the five calcium and phosphorus-deficient rats given viosterol throughout gestation came to term; five of seven given viosterol the last 10 to 14 days delivered young. Other than size and calcium and phosphorus ash content of the pups, no data were given as to their status at birth. The pups born to viosterol supplemented dams, whether or not they had had calcium or phosphorus deficiency, were larger and had higher calcium and phosphorus contents than did control pups or those on the basic diet.
Abnormalities in magnesium metabolism during pregnancy (as a result of, or a contributory factor in, vitamin D, PTH, CT, calcium, and phosphorus imbalances) have been shown to influence profoundly the success of gestation and the status of the newborn infant. Forfar (1976) has listed some of the mechanisms that can con tribute to disturbances in mineral metabolism in the perinatal period. He cited:
1. Inherent (genetic) defects in the parents transmitted to the offspring.
2. Congenital absence or hypoplasia of the parathyroids.
3. Disturbance of the maternal (intrauterine) mineral status with reciprocal fetal disturbances.
4. Nutritional deficiency.
5. Placental insufficiency and IUGR.
6. Prematurity.
7. Perinatal asphyxia and birth injury.
8. Excess phosphorus in infant feedings.
9. Eclampsia.
This listing is useful as a summation of many factors that have been presented in this section, interactions among which are frequent. Some additional data in several of the categories might further explicate some of the interrelationships, and shed some light on metabolic aberrations that might be contributory to clinicopathologic findings in the perinatal period.
3.5.1. Genetic Hypoparathyroidism
Absence or hypoplasia of the parathyroids is usually characterized by symptoms and signs of hypocalcemia. Although often also present, hypomagnesemia is less frequently sought and detected. This disorder is often associated with other endocrinologic abnormalities, including that of the thymus, and by lymphopenia and other immunologic deficiencies. This constellation of abnormalities is suggestive that magnesium deficiency might be an underlying factor, since it causes not only parathyroid dysfunction (Review: Nusynowitz et al. 1976) but also has been implicated in thymic hyperplasic and immunologic abnormalities (Reviews: Hass et al. 1976/1980; Larvor 1976/1980).
3.5.2. Genetic Hyperparathyroidism
Pregnant women with hyperparathyroidism generally have infants with at least transitory hypoparathyroidism (supra vide). However, familial hyperparathyroidism has been implicated in infants with laboratory or autopsy evidence of hyperparathyroidism. Hillman et al. (1964) reported two siblings with marked parathyroid hyperplasia, who were born to consanguinous parents. The first was detected at autopsy, in association with metastatic calcification and osteoporosis. The second was verified at surgery for subtotal parathyroidectomy. Goldbloom et al. (1972) encountered a second pair of siblings with hyperparathyroidism, with all of the typical characteristics: bone demineralization (with signs of rickets), elevated serum calcium and magnesium, and hypophosphatemia. Both survived subtotal parathyroidectomy: the first at 30 months of age, and the second after the first week of life. Their literature review uncovered nine additional cases in seven families. Not included in their list were two infants who had hypocalcemia and hyperphosphatemia, such as are seen with infantile hypoparathyroidism, but who were found to have parathyroid hypertrophy at autopsy (D. H. Andersen and Schlesinger, 1942). Since these infants had arterial calcification, no early rickets but osteitis fibrosis at the time of death at four months, it is possible that their pseudohypoparathyroidism might have been secondary to magnesium deficiency. The data from the infant reported by Monteleone et al. (1975) supports this speculation. He developed seizures on the ninth day of life and had slightly low serum calcium (7 mg/100 ml) and hyperphosphatemia. After intravenous calcium (which only partially controlled the seizures) his serum magnesium level was 0.8 mg/100 ml. Treatment with parenteral magnesium was more effective. A blood specimen taken three days later was found to have elevated iPTH levels. Thus, pseudohypoparathyroidism is another abnormality that might be related to magnesium depletion.
The maternal and intrauterine magnesium and calcium status has been considered in this section, as influenced by PTH, CT, vitamin D, and by impaired placental function, as well as in Chapter 2. The special role of excessive phosphate and vitamin D during infancy is considered in Section 4.3. The risk of prenatal vitamin D deficiency, as a cause of neonatal rickets, persists in groups with high vitamin D requirements. Whether the vitamin D refractoriness of magnesium deficiency might prove germane to the problem in pregnancy requires investigation.
It has been pointed out that very young (adolescent) mothers-who constitute many of the primiparous mothers with complications of pregnancy and premature or low-birth-weight infants-are at particular risk of poor dietary intake, including magnesium, the average intake of which is low during pregnancy. Young multiparous mothers, particularly those whose pregnancies have been frequent, and mothers of twins or greater multiple births, are also especially prone to magnesium depletion. Mothers with diabetes mellitus (a condition noted to be associated with hypomagnesemia even in the absence of pregnancy) have also delivered infants with subnormal magnesium levels. It has also been found that mothers of infants with neonatal hypocalcemic convulsions (such as have been shown to be associated with hypomagnesemia) are often significantly older, of higher parity, and of lower social class than controls (S. Roberts et al., 1973). Such mothers might be presumed to have been on suboptimal magnesium intakes, and to have been depleting their own magnesium stores with each successive pregnancy.
3.5.5. Eclampsia
Of particular importance are the low magnesium levels and high percentage retentions of pharmacologic doses of magnesium given to preeclamptic and eclamptic women. As has been discussed, the high fetal mortality of infants of eclamptic women is being increasingly attributed to placental damage, with resultant intrauterine malnutrition and hypoxia. Brash (1949) reviewed the literature to that time and evaluated 120 full-term live-born infants of toxemic mothers as compared with the same number of infants born after normal pregnancies. The incidence of abnormal lethargy, sometimes with edema or convulsions for days after birth was 11:1 in infants of toxemic mothers versus those born of normal mothers. Stillbirths and neonatal deaths occurred in 10.7 and 5.2%, respectively, of the infants born after eclampsia, and in 3.9 and 2.9% of those born after normal pregnancies. The observation that fetal salvage is improved in eclamptic women treated with magnesium sulfate alone, as compared with that of those given other antihypertensive and anticonvulsant medications (Zuspan and Ward, 1965; Zuspan, 1969) is further suggestive evidence of the importance of magnesium for both mother and infant.
Part I: Chapter 4
MAGNESIUM DEFICIENCY DURING GESTATION, INFANCY, AND EARLY CHILDHOOD
The magnesium levels at birth (indicated by cord levels) reflect the fetal response to maternal conditions during gestation: systemic and placental, and the ease or difficulty of delivery with resultant normal or hypoxic state of the newborn infant. Conditions that lead to neonatal hypermagnesemia might mask an underlying magnesium deficiency. Hypermagnesemia might result from administration of pharmacologic doses of magnesium to the mother shortly before delivery for management of toxemia of pregnancy, or from egress of magnesium from the tissues of infants subjected to anoxia, acidosis, or surgery. Exchange transfusions with citrated blood profoundly affect magnesium as well as calcium homeostasis. Levels during the first week of life reflect the infant's adjustment to independent life in the absence of immediate maternal blood-borne factors, and are affected by the nature of milk and supplements provided. The nature of feeding also influences levels later in infancy. Metabolic abnormalities that interfere with magnesium absorption or retention, although not common, have produced severe mineral imbalances that have focused pediatricians' attention on magnesium. More common conditions, such as severe diarrhea and intestinal malabsorption syndromes, which also cause hypomagnesemia, have further stimulated the pediatrician to be alert to magnesium loss. This section calls attention to the conditions and mechanisms that make infants susceptible to magnesium deficiency and presents speculations as to possible late, as well as overt, immediate sequellae.
4.1. Infantile Magnesium Deficiency: A Factor in Hypocalcemic Tetany, Seizures, and Respiratory Distress
It has long been recognized that neonatal hypocalcemia causes neuromuscular irritability and frank seizures. That the hypocalcemia is secondary to hypomagnesemia in many instances is now clearly established: as a factor in neonatal hypo parathyroidism, in vitamin-D-resistant rickets, and in genetic magnesium malabsorption. Treatment of infantile hypocalcemia with calcemic agents, which can intensify any preexisting magnesium insufficiency, has been shown to cause severe hypomagnesemia and intensification of the clinical manifestations that predicated their use. It is possible that such treatment can be a contributory factor in subsequent renal tubular wasting of magnesium, which can result from intraluminal renal tubular calculi.
Acute magnesium deficiency of infancy severe enough to cause tetany or convulsions, usually in association with hypocalcemia and occasionally with hypercalcemia, was first reported in 1921 by Denis and Talbot. They analyzed plasma calcium levels in 116 hospitalized infants and young children and reported magnesium levels in 38 of those patients. Of the 24 who had hypomagnesemia, six had seizures; two of the older children, four and five years of age, who had been diagnosed as having epilepsy or petit mal had hypocalcemia as well as hypomagnesemia. Three more had tetany; one of those died with laryngospasm at seven months. There were four additional young children (seven months to three years of age) with convulsions, and one with tetany, who had not had their plasma magnesium levels measured. One with microcephaly and mental retardation and one with mental retardation alone had plasma calcium levels of 9.2 and 9.7 mg/100 ml at seven months and two years, respectively. (Another baby with microcephaly and mental retardation, who had plasma calcium of 13.5 mg/100 ml at one year of age, may be the first recorded instance of the infantile hypercalcemia syndrome.) The remaining three babies with seizures or tetany had plasma calcium levels between 5.5 and 8.2 mg/100 ml.
Until the past 15 years, few papers evaluated the magnesium status of infants with abnormalities that later investigations suggest might well have been related to perinatal magnesium deficiency. The infants with tetanic or convulsive signs of hypocalcemia, which were associated with maternal hyperparathyroidism and became worse following treatment with calcemic agents, might have had contributory magnesium deficiency. So, also, might those born after complicated pregnancies or difficult deliveries, which has been shown to predispose to infantile convulsions (S. Wallace, 1972).
The role of hypomagnesemia in infantile convulsions has gained increasing recognition since J. A. Davis et al. (1965) reported an infant with hypomagnesemic neonatal fits, born to a mother with chronic malabsorption, and Paunier et al. (1965, 1968b) identified isolated magnesium malabsorption of infancy as a newly recognized genetic disorder. This condition is associated with hypocalcemic tetany and convulsions that require high doses of magnesium for correction. Use of calcium infusions or calcemic agents, such as high doses of vitamin D or parathyroid hormone, can intensify the neuromuscular irritability, and often do not even correct the hypocalcemia. However, far more infants than those unusual children with magnesium malabsorption are subject to hypomagnesemia. For example, the same year that Paunier et al. (1965) published their preliminary report, Davis et al.(1965) reported an infant boy with convulsions that started on the eighth day of life, and who had hypocalcemia, hypomagnesemia, and hypoglycemia. His intermittent fits became continuous following glucose and calcium infusions that raised his blood glucose to normal but exerted no influence on the hypocalcemia (Fig. 4-1). The seizures stopped within 30 seconds of intravenous administration of 2.5 mEq of magnesium, and his strongly positive Chvostek's sign became negative. The authors considered maternal hyperparathyroidism (secondary to long-term intestinal malabsorption) to have resulted in transitory suppression of her baby's parathyroid function. He responded to PTH by increased clearance of phosphate and decreased calcium and magnesium excretion, despite which his serum magnesium again declined, but without recurrence of convulsions.
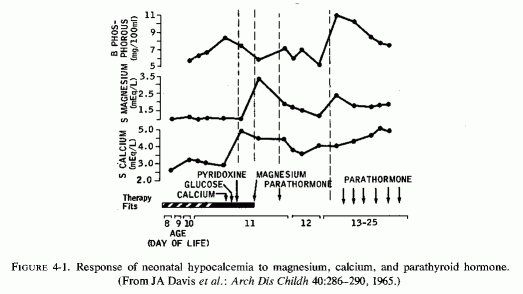{kind=link}
Following the detailed study of the second reported (male) infant with magnesium malabsorption (Salet et al., 1966), and the suggestion that the disease might be hereditary in a third boy (M. Friedman et al. 1967), two more male infants developed convulsive hypomagnesemic hypocalcemia. One was born to a mother with poorly controlled diabetes mellitus (Clarke and Carré, 1967) and thus might have had intrauterine magnesium deficiency. The other was born to a mother with hypophosphatemia, who had received Dilantin therapy for many years (Dooling and Stern, 1967), and thus might have been magnesium deficient before and after birth. The infant born to the diabetic mother (Clarke and Carré, 1967) had had a low Apgar score at one minute and developed respiratory distress a few hours after birth. He had clonic convulsive movements on day 13, which responded to addition of calcium chloride to his formula until day 32, when his convulsions recurred. They intensified on addition of AT-10 (a dihydrotachysterol), high dosage vitamin D, and intravenous calcium gluconate, which did not increase his serum calcium levels. His serum magnesium was then measured and found to be 0.6 mEq/liter. A single intramuscular injection of magnesium (1 ml 50% MgSO4 resulted in cessation of convulsive movements a few minutes after the injection; the improvement persisted thereafter and no further magnesium supplements were given. The infant who had received the exchange transfusion (Dooling and Stern, 1967) showed continuation of irritability, tremulousness, and convulsions, after a focal seizure on day 6, that persisted (during calcium therapy) until his hypomagnesemia was detected and corrected. Atwell (1966) presented detailed studies of three infant boys who developed hypomagnesemia and hypocalcemia and were unresponsive to calcium infusions after neonatal gastrointestinal surgery, but who responded to magnesium (Fig. 4-2).
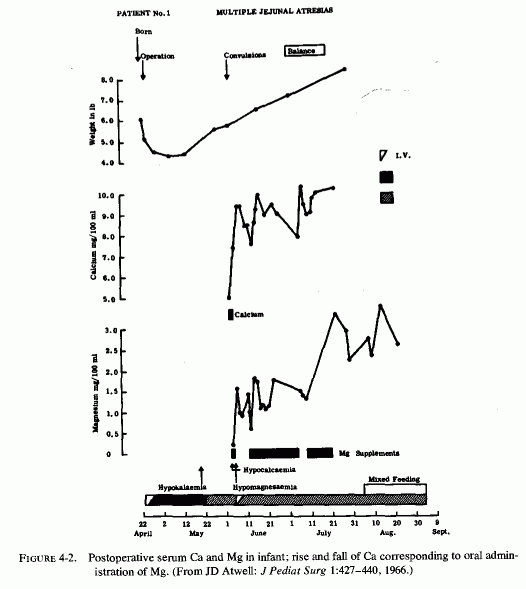{kind=link}
The clustering of reports of neonatal infants, whose hypocalcemic convulsions could be directly attributed to magnesium deficiencies of different etiologies led to an editorial (Canad MAJ, 97:868, 1967) that pointed out that hypomagnesemia is more likely to be a crucial medical problem than a chance occurrence. Stressed was the need for ready availability of facilities to monitor serum magnesium levels, certainly in convulsing infants, and also in other conditions associated with hypomagnesemia, including hypervitaminosis D and use of diuretics, and in the protein-calorie-malnutrition syndrome. Because of sudden death occurring in infants receiving exchange transfusions, and the evidence that citrated blood lowers blood magnesium levels (Bajpai et al., 1967a,b), the editor also called for determinations of magnesium levels in such infants, or preferably using heparinized rather than citrated blood for exchange transfusions. Neonatal infants requiring major surgery, who also generally are transfused, are also at risk of hypomagnesemia (Atwell, 1966; Jalbert et al., 1969).
There have been many published case reports and reviews published since, in which hypomagnesemia is the common denominator in several otherwise unrelated conditions characterized by neonatal and later infantile tremors, tetany, and convulsions. Most are associated with hypocalcemia, but several show a poor correlation with plasma calcium levels. Whether hypocalcemic tetany or convulsions associated with normal magnesium levels in the serum (which can rapidly attain normal levels despite tissue deficit) is another manifestation of a related metabolic disorder requires further study.
The group in Scotland that considers disturbed magnesium metabolism to play a significant role in neonatal convulsions in otherwise normal infants (Forfar et al., 1971/1973; J. K. Brown et al., 1972; Cockburn et al., 1973; Forfar, 1976; T. Turner et al., 1977) observes that this syndrome occurs in bottle-fed, but generally not in breast-fed, infants. They have presented evidence that both plasma and cerebrospinal fluid (CSF) levels of magnesium and calcium are lower in convulsing than in normal infants; the CSF phosphorus level of convulsing infants is normal despite hyperphosphatemia. The babies with convulsions are described as classically "jittery." They found the syndrome to be severe in 35% and lesser in degree more frequently. Among 75 consecutive newborn infants with convulsions considered due primarily to disordered mineral metabolism, seen over a two-year period, subnormal calcium levels (more than 2 S.D. below the mean) were seen in 92%, subnormal magnesium levels in 52%, high phosphorus levels in 67%, and combinations of biochemical disturbances in 80% (Fig. 4-3). Hypocalcemia was associated with hyperphosphatemia in about 60% and with hypomagnesemia in about half of the cases. Hypomagnesemia without hypocalcemia was seen in 7%, almost half of whom also had normal phosphorus levels. Convulsions considered due primarily to brain damage (in 60 additional infants) often also exhibited mineral metabolism derangement, predominantly hypocalcemia and hyperphosphatemia (J. K. Brown et al., 1972). Infants fed evaporated milk formulas had low magnesium and high phosphorus levels, comparable with levels of convulsing infants in 68 and 80% of the controls. In an evaluation of clinical and chemical relationships in neonatal convulsions, the group (J. K. Brown et al., 1972) commented that they had encountered convulsions in 1.4% of live-born infants. Most of those classified as due to brain damage occurred in the first three days of life; most of those considered metabolic in origin occurred from the fourth day on (Fig. 4-4). They noted that the proportion of metabolic to brain-damage convulsions seems to have risen markedly in reports published since 1969, as compared with reports published between 1954 and 1960, during which time brain-damage-induced convulsions were predominant. Since metabolic convulsions are more amenable to correction, this is an important point in terms of management of convulsing infants.
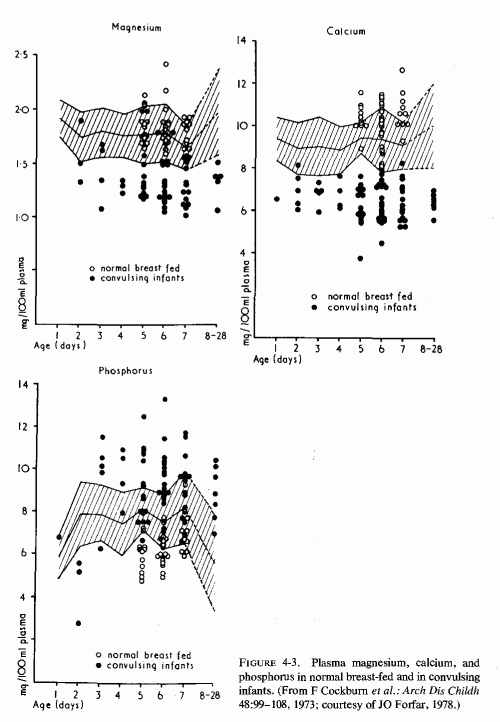{kind=link}
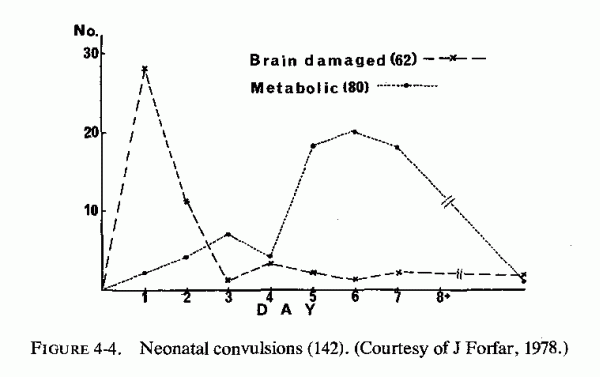{kind=link}
Wong and Teh (1968) had earlier reported hypomagnesemia without hypocalcemia in five otherwise normal infants, during the week after birth. (This was part of a study of 40 babies and young children with convulsions, tremors, or muscular twitchings, 13 of whom had hypomagnesemia alone, and 27 of whom also had hypocalcemia). When symptoms were present, both total and ultrafiltrable mean levels of magnesium were significantly lower than in controls (p = < 0.001). The major decrease was in the ultrafiltrable moiety. Keen (1969), like Forfar and his colleagues (supra vide) called attention to the increasing incidence of infantile convulsions of metabolic origin in England. Of 100 infants with seizures in the first 4 weeks of life, 36 had hypocalcemia, with peaks of incidence in the first 48 hours and between the 4th and 10th days of life. Only toward the end of this 23-month study were magnesium levels determined. Details of the inconstant association of hypomagnesemia with hypocalcemia were not given, but the investigator considered the response of refractory hypocalcemic fits to magnesium (Davis et al., 1965) as suggestive of its importance in this syndrome. He, too, commented on the disproportionate distribution of convulsions among bottle-fed as compared with breast-fed infants. Harvey et al. (1970) also showed that the mean magnesium level was lower in bottle-fed than breast-fed infants by the seventh day of life, and that among those with convulsions the mean was even lower. In this series, even many of the nonconvulsing infants had hypomagnesemia and hypocalcemia. This recalls Bruck and Weintraub's (1955) admonition that asymptomatic hypocalcemia should not be considered "physiologic," since transition from latent to manifest tetany is frequent and can occur unexpectedly. The same is likely to be true for hypomagnesemia. Furthermore, because of the evidence that prolonged chronic magnesium deficiency can contribute to cardiovascular, renal, and bone abnormalities, overt symptomatology may not be the major risk.
The infant reported by Vainest et al. (1970) might be an example of delayed as well as acute complications of magnesium deficiency of infancy. Although this infant had not had his severe hypomagnesemia (0.4-0.7 mEq/liter) detected until three days before he died at five and a half months, there is strong inferential evidence that magnesium deficiency was likely to have played a contributory role. He was the ninth child of a woman who had been treated for tuberculosis, and thus was probably magnesium depleted. [High parity contributes to the magnesium drain on the mother, and aminoglycoside antibiotics are magnesium wasters (Vanasin et al., 1972).] Five of her seven sons had had seizures; three died. Two, counting the propositus, whose hypomagnesemia had been identified late (after massive calcemic therapy), had arterial calcinosis. That infant also had renal and myocardial calcinosis.
The frequency of low magnesium levels among infants with symptomatic hypocalcemia was noted by the investigators cited above, and in subsequent studies. Stern and Harpur (1971/1973) briefly reported six newborn infants whose hypocalcemia was clearly secondary to their hypomagnesemia. Radde et al. (1972) commented that symptoms and signs attributable to low ionized calcium levels were found only in infants who had low plasma levels also of magnesium. Tsang (1972), who reviewed in detail the factors contributing to neonatal magnesium disturbances, also commented on the concomitant hypocalcemia, and vice versa. Subsequent work from his group has elucidated the infants at greatest risk of the combined divalent cation deficiencies (Tsang et al., 1973, 1974, 1976, 1977a,b; Tsang and Brown, 1975, 1977). David and Anast (1974) found that plasma magnesium levels were significantly lower in hypocalcemic than in normal or sick neonates.
Most of the infants described in this section were newborn. Convulsions and tetany associated with hypomagnesemia have also been reported in older infants and young children. Febrile convulsions are frequently associated with lower than normal serum magnesium levels, often without hypocalcemia (Chhaparwal et al., 1971). A "meningo-encephalitic, or tremor" syndrome in Indian children has also been associated with hypomagnesemia in infants of 6-24 months of age, who have evidence of mental retardation and malnutrition (Chhaparwal et al., 1971/1973). Severe magnesium deficiency also occurs during repair of protein calorie malnutrition (see pp. 122-128).
4.1.2. Low-Birth-Weight Infants
Lower cord blood magnesium levels have been reported in low-birth-weight infants than in full-term infants (Breton et al., 1960; Review: Ferlazzo and Lombardo, 1971). When the low birth weight is due to prematurity, the low cord blood levels can be attributed to the subnormal accumulation of minerals in the final weeks of gestation. Widdowson and Dickerson (1962), who have tabulated the mineral contents of 1.5 kg, 2.5 kg, and full-term babies, have shown that the magnesium content of the more immature or smaller babies is only 42% that of normal size infants, while that of 2.5-kg infants is 76% that of the normal full-term baby. In regard to the tendency toward hypocalcemia of premature infants, the 1.5- and 2.5- kg infants have 36% and 68% the calcium contents, respectively, of full-term babies. This study did not differentiate between immature infants and those that are small for gestational age (SGA) as a result of intrauterine growth retardation (IUGR).
The hypocalcemia and hypomagnesemia of low-birth-weight infants can reflect inadequate stores accumulated before birth, in addition to postnatal problems in homeostasis. Their hyperphosphatemia can derive from tissue breakdown and be aggravated by inappropriate dietary intakes, functional immaturity, and hormonal imbalances. The hyperphosphatemia associated with hypocalcemia and hypomagnesemia that is found in full-term infants fed cows' milk rather than breast milk and that is aggravated by vitamin D is further discussed on pp. 105-108.
Renal tubular immaturity has been proposed as an explanation of the inability of the neonate to eliminate excess phosphate, whether endogenous or exogenous, that is associated with persistent hypocalcemia and hypomagnesemia. Rubin et al. (1949) showed that aspects of renal function mature at different rates, usually reaching adult values during the second year of life. Dean and McCance (1948) and L. Gardner et al. (1950) reported that renal tubular immaturity was responsible for the low phosphate clearance that they reported in neonatal infants. This fits the experimental evidence suggesting absence of fetal phosphaturic response to exogenous PTH (Garel and Barlet, 1974). Tsang et al. (1973b) found that phosphorus excretion increased in premature infants over their first three days of life, whether or not PTH was given. Their fractional tubular reabsorption fell and there was no significant difference in phosphorus excretion or reabsorption between the PTH-treated and nontreated infants.
The theory that transient hypoparathyroidism of infancy is a result of parathyroid immaturity has been discussed earlier. If valid, this theory is even more applicable to low-birth-weight infants and might explain their subnormal PTH response to neonatal hypocalcemia. Also suggested frequently is the possibility that fetal hypercalcemia, possibly deriving from maternal hyperparathyroidism-induced hypercalcemia, might cause fetal PTH suppression, mediated by resultant fetal hypercalcemia (Review: Tsang et al., 1976b). However, cited experimental studies have shown that experimental dietary- or hyperparathyroidism-induced calcium and phosphate aberrations are not reflected by parallel changes in the fetal blood. Fetal parathyroids function to maintain the calcium homeostasis. Furthermore, hypercalcemia during late gestation is uncommon even in the presence of "physiologic" hyperparathyroidism. Thus, it seems plausible that it is not parathyroid immaturity but postnatal factors that prevent normal PTH reactivity. For example, hypocalcemic hyperphosphatemic premature infants have responded to injections of exogenous PTH with transient rises in serum calcium and magnesium in the first few days of life (Tsang et al., 1973; David and Anast, 1974; Rootet al., 1974), indicating that there was bone mineral mobilization in response to PTH (Fig. 4-5, Tsanget al., 1973a), even in infants born prematurely.
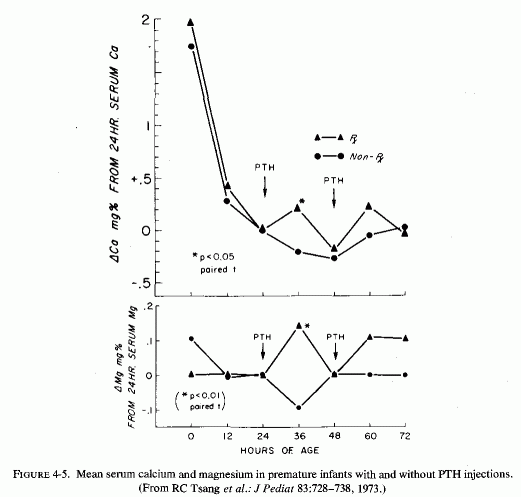{kind=link}
In the case of infants with IUGR, such as are commonly born to mothers with toxemias of pregnancy and to young primiparous mothers, significantly lower levels of serum magnesium have been detected than in other low-birth-weight infants (Fig. 4-6, Tsang and Oh, 1970: Jukarainen, 1971). Tsang and Oh (1970) suggested that the low serum magnesium levels in IUGR infants might reflect disturbed placental transfer of magnesium or abnormal fetal magnesium metabolism as part of the intrauterine malnutrition syndrome. Hypocalcemia has been shown to be more striking than hypomagnesemia in IUGR neonates (Tsang et al., 1975). Such neonatal hypocalcemia in infants with placental insufficiency has been associated with impaired transfer of calcium from mother to fetus (Khattab and Forfar, 1971). Tsang et al. (1975) suggest that their findings (Tsang et al., 1973a,b, 1974) point toward shortened gestational age or birth asphyxia as more likely explanations of the disturbances in calcium homeostasis during the early neonatal period. The greater tendency of IUGR infants than full-term infants to have poor bone mineralization and spontaneous bone fractures suggests that maintenance of divalent cation homeostasis in utero might be achieved by hyperactivity of fetal parathyroids in response to intrauterine malnutrition, when there is faulty placental transport of calcium and magnesium from maternal to fetal circulation.
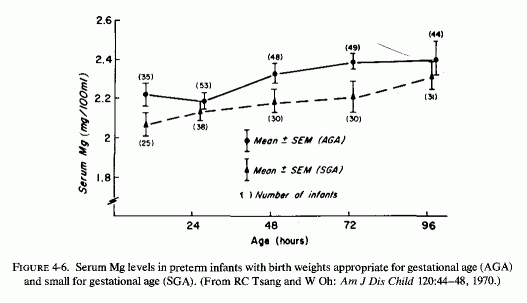{kind=link}
The observation that IUGR infants often exhibit neonatal hyperirritability and jitteriness (Michaelis et al., 1970; Ferlazzo and Lombardo, 1971; Tsang et al., 1975) suggests that, in addition to hypocalcemia, magnesium deficiency also be considered. The failure to find hypomagnesemia at 4 hours, and its decline by 24-48 hours, especially in infants whose hypocalcemia also becomes more notable at that time (Fig. 4-7, Tsang et al., 1975b), suggests that hypoxia at birth, which is common in IUGR infants (Tsang et al., 1975), can be contributory and might mask the magnesium deficiency. Serum magnesium values being a poor index of tissue magnesium status, percentage retention of magnesium-load tests might prove a more valid means of ascertaining whether the irritability of IUGR infants can be partially attributed to magnesium deficiency (Harris and Wilkinson, 1971; Caddell, 1975).
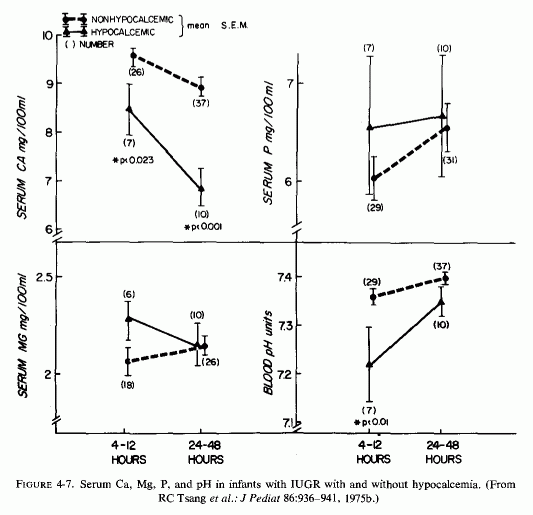{kind=link}
4.1.3. Neonatal Hypoxia
Infants born after difficult deliveries and who have birth apnea have been found to have hypermagnesemia shortly after birth (Engel and Elm, 1970). It is probable that the source of this elevated serum magnesium is from the tissues, injured as a result of the hypoxia, as has been demonstrated in war injuries and clinical or experimental shock (Beecher et al., 1974; Root et al., 1947; Canepa and Gomez-Pavira, 1965; W. Walker et al., 1968; N . Goldsmith et al.,1969; Flynn et al., 1973, 1976/1980). The accompanying acidosis enhances the shift of bone minerals to the extracellular space (Barzel and Jowsey, 1969; Raisz, 1970). Thus, such infants, despite their transient hypermagnesemia or normal magnesium levels (Fig. 4-8) (Tsang et al., 1974), may actually suffer from body depletion of magnesium. Their drop in serum calcium in the first few days of birth has been generally blamed for the hyperirritability, jitteriness, convulsions, and periods of apnea, common in hypoxic infants (Oppé, 1970). However, they frequently also show as striking depressions in their serum magnesium levels and a lesser drop in serum phosphorus (Fig. 4-9, Tsang et al., 1974). The rise in serum phosphorus, which precedes the rises in the divalent cations, suggests that PTH-mediated mobilization of bone mineral might not then be operative. The rise in serum phosphorus can be caused by several factors. The initially higher than maternal values might be endogenous in that it is caused by endogenous tissue breakdown, which is associated with stress of delivery and birth asphyxia. The subsequent rise might derive from bone mineral efflux, high phosphate intake (from cows' milk), and renal tubular inability to eliminate the phosphorus load in the early days of life. Asphyxiated infants, whose serum magnesium levels dipped only slightly at 12 hours and then rose to normal by 24 hours, were compared with asphyxiated infants whose hypoxemia (starting at 12 hours) persisted through 48-72 hours (Tsang et al., 1974). The hypocalcemia of the latter group was more profound, and correction of acidosis took longer than it did in the asphyxiated infants with normal serum magnesium levels. The drop in serum magnesium levels within 12-24 hours after asphyxia may well reflect the low reserves of magnesium in neonatal infants, or the shift from extracellular to intracellular space on correction of the hypoxia and acidosis.
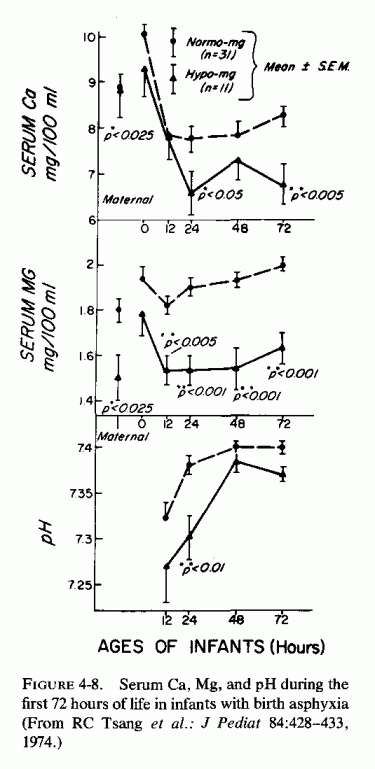{kind=link}
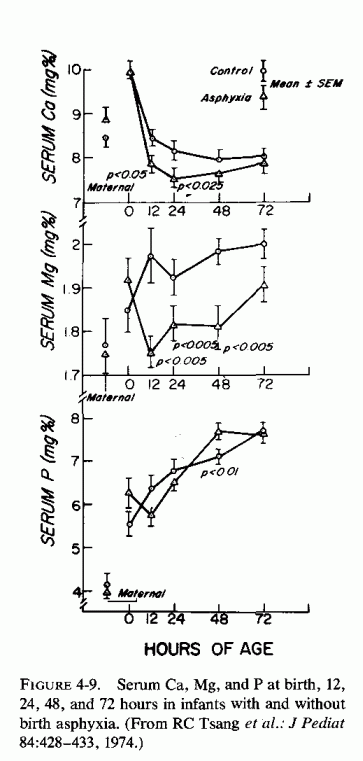{kind=link}
Infants of diabetic mothers can either be premature or large for gestational age, often exhibit respiratory distress and acidosis, and also frequently show rising serum phosphorus and falling serum calcium and magnesium levels by 24-48 hours after birth (Fig. 4-10, Tsang et al., 1972). This had been speculated to reflect maternal hyperparathyroidism of diabetic mothers. However, Tsang et al.(1972) noted that diabetic mothers had serum calcium levels within normal limits. Since they did not have hypercalcemia, suppression of fetal parathyroids from this source seems questionable. Functional hypoparathyroidism of the infants was considered unlikely when they were found to exhibit short-term calcemic response to PTH injections (Fig. 4-11), indicating bone mineral mobilization. Although administration of PTH to infants of diabetic mothers caused more phosphaturia than was seen in nontreated infants of diabetic mothers, there was no difference in percentage tubular reabsorption of phosphorus in the two groups, suggesting renal immaturity. Their subsequent work showed no significant difference in serum PTH or total or ionized calcium levels in diabetic than in normal mothers (Tsang et al., 1975). Since they found that PTH levels of cord blood of infants of diabetic mothers (IDM) were not significantly lower than were those of controls, they assumed that the parathyroids of the IDM functioned as did those of normal infants. The observation that there was no significant increase in PTH levels in response to significant decreases in total and ionized calcium led Tsang et al. to assume a failure of production of PTH. Prematurity (9 of 13 infants of insulin-dependent mothers with gestational ages of 37 weeks or less), birth asphyxia (10 of the 28 IDM had 1 minute Apgar scores of 6 or less), and increased calcitonin secretion were also considered as possible explanations for the sustained hypocalcemia of the infants of diabetic mothers. The changes in IDM serum magnesium were not considered significantly different from those of controls in that study. However, although the maternal serum magnesium levels were within the same range in control and diabetic mothers, it is of interest that the cord blood levels of the normal infants, which were low, rose to about 1.7 mEq/liter by 76-96 hours, whereas the mean values of infants of insulin-dependent mothers remained about 1.5 mEq/liter. Their range of values at 24-48 hours was 1.35-1.7 mEq/liter and at 72-96 hours was about 1.4-1.5 mEq/ liter. The following year, Tsang et al.(1976b) reported that 21 of 56 IDM had serum magnesium levels at or below 1.25 mEq/liter on at least one occasion during the first three days, and that they did not exhibit the normal increase with postnatal age seen in normal infants. Subnormal neonatal serum magnesium levels were related to the degree of severity of diabetes, youth of the mothers, lower gravidity, and prematurity. Lower concentrations of serum magnesium were associated with less increase (or actual decreases) in serum concentrations of PTH from 48-72 hours, and conversely serum concentrations of magnesium at 72 hours were related to parathyroid function from birth to 24 to 48 hours of age (Fig. 4-12, Tsang et al., 1976c). Since diabetes mellitus is recognized to cause magnesium deficiency without the added requirements caused by pregnancy, it is not surprising that infants of diabetic mothers are particularly subject to magnesium deficiency. The interrelationship of their magnesium inadequacy, phosphate excess, and hypocalcemia with their parathyroid malfunction is an important clue to the complex hormonal/mineral interrelationships that may be mediated by a fundamental magnesium deficit.
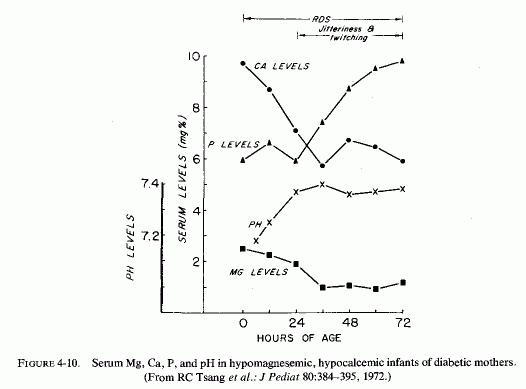{kind=link}
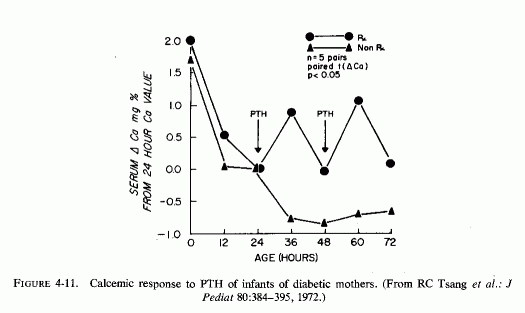{kind=link}
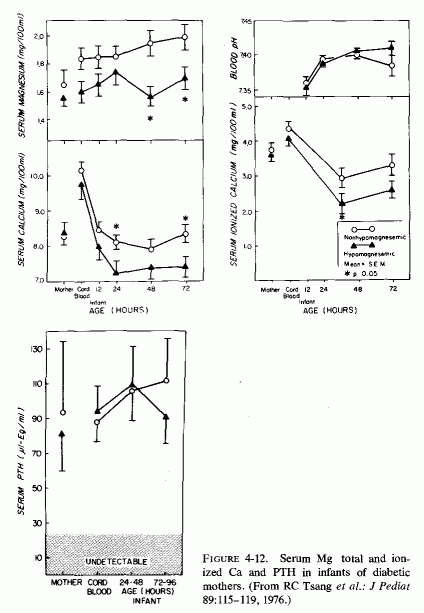{kind=link}
4.1.5. Neonatal Hypermagnesemia
Hypoxia has been shown to cause loss of magnesium from tissues with resultant elevation of serum magnesium levels. Studies of serum from venously occluded arms (Whang and Wagner, 1966; S. P. Nielsen, 1969) have shown that even short periods of hypoxia cause egress of magnesium from the cells to the blood. Thus, it is not surprising that infants born after difficult deliveries and with birth asphyxia have had elevated serum magnesium levels at birth and shortly thereafter (Engel and Elm, 1970; Donovan et al., 1977b). Such infants, however, often exhibit hypomagnesemia within 12 hours after birth (Tsang et al., 1974), possibly reflecting inadequacy of tissue stores or the shift of extracellular magnesium to the intracellular phase with normal oxygenation.
Acidosis, common in low-birth-weight infants, is another cause of neonatal hypermagnesemia. Even minor drops of muscle pH (to 6.8) has been shown in vitro to cause significantly decreased muscle magnesium content (Gilbert, 1961). A clinical reflection of this observation is the hypermagnesemia of decompensated diabetic acidosis (Marlin et al., 1958). Thus, the normal or elevated serum magnesium seen in acidotic infants immediately after birth, despite the evidence that such infants are at risk of hypomagnesemia, should come as no surprise. Even normal infants have acidosis, due to elevated maternal lactic acid levels and to the period of anoxia during birth (Acharya and Payne, 1965). The levels fall as oxygenation is established, normally reaching adult values after two days. Infants with respiratory distress have prolonged acidosis and anoxia, which militate against restoring tissue levels of magnesium. This set of circumstances is likely to mask the underlying magnesium deficiency when serum magnesium levels are relied upon to reflect the magnesium status.
The most intensive study found on the magnesium levels of the neonate (Jukarainen, 1974) demonstrates that high-risk infants with hypocalcemia (whom one would expect to have hypomagnesemia) are likely to have normal magnesium levels in the early hours to days after birth. This investigator correlated many factors that influence neonatal homeostasis, considering gestational and perinatal abnormalities. As many as nine blood samples were analyzed for Mg/Ca/P in the infants during the first five days of life. He found that these longitudinal studies showed that there was an inverse correlation between the serum magnesium and gestational age. The premature and low-birth-weight infants (who have been shown to be more susceptible to hypocalcemic tetany and convulsions) had essentially normal serum magnesium with their hypocalcemia in the first five days, as compared with full-term infants whose hypocalcemia correlated positively with hypomagnesemia during the same period. Infants of diabetic mothers also showed relatively higher serum magnesium levels, in association with their hypocalcemia during the first few days, but the magnesium levels tended to drop toward the end of the observation period. Jukarainen (1974) concluded that the inverse relationships between calcium and magnesium levels in the early days of life of the high-risk infants probably reflected disturbed magnesium homeostasis (such as has been seen with hypoxic and acidotic egress of magnesium from the cells).
Direct evidence that this might explain the above findings was provided by Yamashita and Metcoff (1960), who found that the skeletal muscles of premature infants were edematous, and that the levels of normal intracellular cations and of magnesium-dependent enzymes were significantly lower than normal. Chiswick (1971) also noted edema in hypocalcemic neonatal infants, and noted that the serum magnesium levels of the hypocalcemic infants were higher in infants with edema than in those without.
Infants born to mothers given pharmacologic doses of magnesium for eclampsia shortly before delivery have been born with hypermagnesemia and secondary respiratory depression, areflexia, and paralysis (Fishman, 1965; Brady and Williams, 1967; Lipsitz and English, 1967; Lipsitz, 1971). Serum levels as high as 15 mEq/liter were detected in one such infant, who recovered following treatment by exchange transfusion (Brady and Williams, 1967). However, Lipsitz (1971) found no correlation between (1) the cord or newborn serum magnesium levels and the Apgar score; (2) the total dose of magnesium given to the mother and her serum magnesium level at delivery, or that of the cord blood; and (3) the total dose of magnesium and clinical evidence of neonatal magnesium toxicity.
Unlike adults, who excrete infused magnesium rapidly (Chesley and Tepper, 1958), neonates have a very low magnesium excretion rate (Lipsitz, 1971; Tsang, 1972). During the first few days of life, glomerular filtration rates are low (less than 0.34 mg/kg/24 hours); in premature infants the glomerular filtration rate and magnesium excretion is even less than in full-term infants (Tsang, 1972). Thus, it is not surprising that it has taken up to five days for neonatal hypermagnesemia to fall to normal levels (Lipsitz, 1971). Despite sustained elevated serum magnesium levels in infants born to toxemic mothers, given large amounts of magnesium for different periods of time before delivery, there have been surprisingly few instances of serious manifestations of hypermagnesemia. For example, only 8 of the 118 infants born to mothers given 30-40 g of magnesium sulfate i.m. during the 24 hours before delivery, had Apgar scores of 5 or less; none had cord magnesium levels above 6 mEq/liter during labor; no detectable adverse effects attributable to the magnesium alone were detected (Hutchinson et al., 1963).
The meconium plug syndrome, attributed to hypermagnesemic suppression of peristalsis, has been reported in two infants born prematurely to two eclamptic young women given high-dosage magnesium therapy shortly before delivery (Sokal et al., 1972). The cord blood serum magnesium level was 8.3 mEq/liter in the infant from whom it had been obtained; it was 6.0 mEq/liter at 3 hours of age in the other. It had dropped to 5.4 mEq/liter by 6 hours, 4.3 at 55 hours, to 4.3 mEq/liter in the first infant, and to 4.2 mEq/liter at 10 hours in the second. Neither had hypocalcemia at any time tested. Since epsom salt enemas have been known since the turn of the century to cause magnesium toxicity in children and adults (C. Fraser, 1909; Fawcett and Gens, 1943), this treatment of hyaline membrane disease, which has led to fatal consequences of severe hypermagnesemia, is no longer recommended (Tsang, 1972; Outerbridge et al., 1973).
Exchange transfusions with blood to which acid-citrate-dextrose (ACD) solution has been added are known to cause infantile hypomagnesemia (Dooling and Stern, 1967; Bajpai et al., 1967a,b; Z. Friedman et al., 1971,1972). Although it has long been known that weakly dissociated salts of citrate are formed with both magnesium and calcium (Hastings et al., 1934; Walser, 1961), and citrate infusions to dogs have caused both hypomagnesemia and hypocalcemia [total (Bunker et al., 1962) and ionized (Killen et al. 1971)], the customary procedure for infants receiving exchange transfusions who develop irritability, seizures, or cardiac arrhythmias (Dooling and Stern, 1967; Rosefsky, 1972) has been to provide calcium with the transfusion and to monitor the serum calcium levels. Generally, only when the condition fails to improve has the magnesium status been explored and magnesium therapy instituted. An editorial (Canad MAJ, 97:868, 1967) considered sudden death during the course of the exchange a possible consequence of the citrate-induced reduction in serum ionic magnesium. Two groups of investigators in Canada demonstrated that the serum ionic magnesium dropped substantially during exchange transfusion with ACD blood (Bajpai et al., 1967a,b; Z. Freidman et al., 1971, 1972). The first group (Bajpai et al., 1967b) noted electrocardiographic changes (flattening of T waves) when the serum Mg fell below 0.8 mEq/liter. The second group (Z. Friedman et al., 1972) considered it likely that the magnesium-responsive arrhythmia that developed during the fourth ACD plus calcium transfusion (Rosefsky, 1972) was likely to have reflected also a reduction in ionic calcium, despite the administration of calcium gluconate. A more detailed report (Radde et al., 1972), presented evidence that abnormal symptoms and signs are found almost exclusively in infants whose plasma levels of both cations were below the lower limits of normal. Recently Donovan et al. (1977a) showed that exchange transfusion (which they confirmed lowered serum levels of ionized magnesium and calcium) increases PTII levels, as measured by immunoassay.
An overload of citrate similar to that of the exchange transfusion of infants is the use of ACD blood prime in cardiopulmonary bypass procedures. Killen et al. (1972) showed that severe depression of ionized magnesium (Fig. 4-13A) could be prevented by adding magnesium sulfate: 3 ml of 10% solution per unit of ACD blood (Fig. 4-13B). Since low magnesium levels are common in patients to undergo open- heart surgery, magnesium therapy of such patients is often necessary (Holden et al., 1972; Khan et al., 1973).
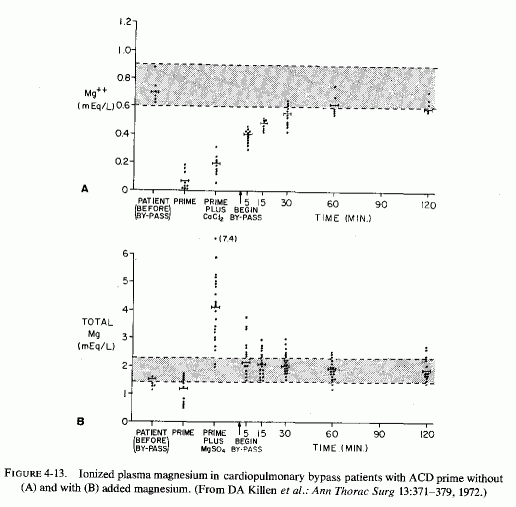{kind=link}
When transfusions, using citrated blood, are given to those whose underlying condition makes, it likely that they might be magnesium deficient before the transfusion, severe depletion may ensue. Jalbert et al. (1969) reported such an instance in the case of a premature infant born to a preeclamptic mother. The infant developed mucoviscidosis and intestinal obstruction requiring resection, during which citrated blood transfusions were given. Calcium-refractory seizures developed that responded only to magnesium repletion.
In view of the drop in ionized calcium and magnesium caused by citrated blood transfusions, attention should be paid to other more common conditions in which neonatal tetany has been correlated with decreased ionized calcium levels. (Ionized magnesium is less readily measured, and thus is rarely reported.) The possibility that asymptomatic neonatal hypocalcemia might be related to normal levels of ionized calcium despite low total calcium has long been suspected (Bruck and Weintraub, 1955) and more recently verified. Bergman (1972) showed that symptomatic neonatal hypocalcemia is associated with lower levels of ultrafiltrable fractions of calcium than of total calcium. On the other hand, D. M. Brown et al. (1972) measured the ionized fraction of calcium, and found no correlation between low ionized calcium levels and symptomatic hypocalcemia. Sorell and Rosen (1975) found symptoms with decreases in ionized calcium to a critical level of 2.5 mg 100 ml. Bergman (1974) showed that up to 10-12 hours after birth, the decrease in total calcium is mostly caused by a decrease in the ultrafiltrable fractions. Since symptomatic hypocalcemia seems to be better related to les decreases in ultrafiltrable calcium that consists of ionized calcium plus complexed calcium (about 14% of total calcium: Walser, 1961) than to the ionized fractions, it may be speculated that the change from asymptomatic to overt hypocalcemia might be contributed to by a drop in the complexed fraction. It can be presumed that the HPO4 fraction is unlikely to be low in a condition associated with hyperphosphatemia. The citrate fraction, which is dependent on vitamin D, seems a likely candidate for consideration. It is a complex question, however, since vitamin D deficiency (in rats) has been correlated with decreased blood and bone citrate levels (Harrison et al. ., 1957). Vitamin D administration to rachitic rats has raised the citrate levels (Steenbock and Bellin, 1953), but excess vitamin D (as in acute infantile hypercalcemia related to hyper reactivity to vitamin D) is associated with subnormal blood citrate levels (Forfar et al., 1959; Lindquist, 1962). Radde et al.(1972) found that, at least in newborn infants, symptomatic hypocalcemia only occurred when low ionized calcium levels were present with concomitant hypomagnesemia, an interesting observation in view of the vitamin D resistance of magnesium-deficient patients. Sorell and Rosen (1975), finding both normal and low serum magnesium levels in symptomatic hypocalcemia, did not confirm the report of Radde et al. (1972). However, of the seven infants and young children they reported, all but one (who had sepsis and thus might have had acidosis) had hypomagnesemia. The other two with normal serum magnesium levels in their series of nine were 17- and 19-year-old patients with renal failure, a condition that has been associated with tissue magnesium depletion despite even hypomagnesemia (Lim and Jacob, 1972c). One of the infants developed hypomagnesemia and hypocalcemia after cardiac surgery.
When acidosis develops in the newborn infant, it is customary to treat it with sodium bicarbonate or sodium lactate. Unfortunately, the conditions that give rise to acidosis not infrequently are associated with magnesium egress from the cells. Infusions of sodium lactate cause substantially increased urinary output of magnesium (Barker et al. 1959). Thus, the production of negative magnesium balance in infants whose postoperative acidosis was thus corrected, and the production of hypomagnesemia (Atwell, 1966), is not surprising. (The stress of surgery also increases magnesium loss.) Correction of renal acidosis with lactate, citrate, or bicarbonate has also caused hypomagnesemia (Randall, 1969). Administration of sodium bicarbonate to acidotic neonatal infants has reduced serum ionic calcium levels (Radde et al., 1972; Tsang et al., 1977a,b) and has also lowered serum magnesium levels (Radde et al., 1972; Jukarainen, 1974). The higher the serum bicarbonate levels, the lower the serum magnesium levels (Jukarainen, 1974).
It has been reiterated that infants with hypomagnesemia should not be treated with calcium or vitamin D (Tsang et al., 1977a; Seelig, 1978/1980). Nonetheless, since hypocalcemia is usually detected first in convulsing infants (magnesium determinations often being obtained only on failure of calcemic therapy to correct either the symptomatic or biochemical abnormalities), calcium alone or with vitamin D is still usually the first approach. In fact, prophylactic administration of calcium has been recommended for low-birth-weight or asphyxiated infants who are at particular risk of hypocalcemia (D. R. Brown et al., 1976; Salle et al., 1977). It is realized and cautioned that when symptomatic infantile hypocalcemia is found, hypomagnesemia should be sought (Editorial, Bruit. Med. J., 1973; Tsang et al., l977a). The observation that symptomatic infantile hypocalcemia develops almost exclusively when there is concomitant hypomagnesemia (Radde et al., 1972) lends support to the importance of seeking out a magnesium deficit. Since magnesium is predominantly an intracellular cation, and since levels in the blood are generally kept within narrow limits, relying on serum magnesium as the index of magnesium status of the body can give misleading information. This is particularly true for neonatal infants, whose serum magnesium can be elevated as a result of acidosis or asphyxia-induced egress of magnesium from tissue. The parenteral magnesium-load test is more reliable as a clue to magnesium depletion. For example, Harris and Wilkinson (1971) found that of nine infants suspected of magnesium deficiency, who had serum magnesium levels that were normal, four were deficient by the loading test, Byrne and Caddell (1975) found that there were infants in their survey whose magnesium deficiency would not have been detected by serum levels alone.
With high-risk infants, whose body stores of magnesium might be precariously low, it is possible that treatment directed toward correction only of hypocalcemia might thereby not only fail to correct convulsions, but might intensify occult cardiovascular and renal lesions. Such damage is caused by experimental magnesium deficiency, and is worsened by calcium, phosphate, and vitamin D excesses. Among infants with severe imbalances (low Mg/high Ca, P vitamin D intakes), the damage might be severe enough to cause acute and chronic signs and symptoms during infancy, leading to early death or chronic disorders that might be termed "congenital." Among those with less marked imbalances (i.e., whose prenatal stores were higher or whose postnatal calcemic challenges were less, there might be lesser degrees of damage that might lay the groundwork for adult cardiovascular and renal disease.
It seems likely, even though magnesium determinations had not been made, that the two infants described by D. Andersen and Schlesinger (1942) might have reflected the first of the two possibilities: convulsive hypomagnesemic hypocalcemia treated with calcemic agents, resulting in death in the fourth month of life. In addition to administration of calcium gluconate and moderately to extremely high doses of vitamin D (that lowered, rather than raised, the serum calcium levels) both infants were also given repeated blood transfusions for refractory anemia, and both were treated repeatedly for refractory acidosis. It is conceivable that the anemia was a sign of magnesium deficiency (Elm, 1973, 1976/1980). It is plausible that the calcium- and vitamin-D-refractory hypocalcemic neuromuscular irritability and seizures of both infants might have been caused by early magnesium deficiency that interfered with response to the calcemic agents, and that was intensified by that treatment and by the use of citrated blood for the anemia, and lactate and bicarbonate for the acidosis. One vomited several times daily and developed hypercholesterolemia; the other developed hypertension-all signs of vitamin D excess and in the case of increased blood pressure of a high Ca/Mg ratio. Both had peripheral and coronary arteriosclerosis; one had myocardial infarctions and the other had cardiomegaly. Both had severe renal damage: one predominantly fibrous replacement; the other (who had been given 300,000 IU vitamin D) also had renal calcinosis. Although their biochemical findings suggested hypoparathyroidism, they both had hypertrophied parathyroid glands and bone pathology, and were diagnosed at autopsy as having renal hyperparathyroidism. In view of the data reviewed in the foregoing section, the possibility that these infants had hyperparathyroidism secondary to magnesium deficiency, and that the deficiency interfered with the response of target organs to PTH (pseudohypoparathyroidism) or to vitamin D, and led to cardiovascular and renal disease should be seriously considered. Almost a quarter of a century later, severe hypomagnesemia (0.8 mEq/liter) was correlated with high-dosage vitamin D and calcium treatment of an infant whose hypocalcemic convulsions had started at one month (Salet et al., 1966), as in the prior two cases. Treatment with both cations was then instituted, with resultant elevation of low calcium levels to normal. Both hypocalcemia and hypomagnesemia (0.3 mEq/liter) recurred at three months, after treatment had been stopped. The baby again responded to combined cation therapy. When treatment was again stopped, he exhibited hyperphosphatemia, as well as hypocalcemia. PTH administration corrected the blood calcium and phosphorus levels, but lowered the blood magnesium level (0.5 mEq/liter). Vitamin D therapy again intensified the biochemical abnormalities and the convulsions. Like the infants described by Andersen and Schlesinger (1942) this infant's findings suggested hypoparathyroidism. However, his hypomagnesemia was identified early and treated intermittently until it became manifest that his vitamin-D-resistant hypocalcemia was secondary to magnesium malabsorption. This group found that high-dosage vitamin D increased his magnesium requirements and that treating with both magnesium and calcium was not as effective in raising cellular magnesium to normal levels as was treating with magnesium alone. They later found that this infant's magnesium malabsorption was familial, when a sibling was born with the same defect (Salet et al., 1970). High dosage vitamin D (100,000 IU daily) for familial hypoparathyroidism and convulsive hypocalcemia resulted in hypomagnesemia in a baby from a family with a high incidence of convulsions (Niklasson, 1970). This infant developed emotional lability and mental retardation, similar to that seen with hypervitaminosis D (Review: Seelig, 1969b). Her young sister later also developed hypomagnesemia. It was noted that infantile convulsions, with death during infancy (including one sudden unexplained death at four weeks), were common in the family of these sisters, whose parents were first cousins. The possibility that there was primary magnesium malabsorption or renal magnesium wasting in this family was not explored. The infant son (ninth child of a mentally retarded mother), who developed convulsions after three months of vitamin-D-supplemented (400 IU/day) dried milk formula, was the fifth son to develop seizures (Vainsel et al., 1970). Intravenous calcium gluconate and high dosage vitamin D (750,000 units per week) raised the serum calcium to low normal levels, but failed to control the seizures. Hypomagnesemia (0.4-0.7 mEq/ liter) was then identified, and magnesium therapy was begun three days before death. He had microfocal myocardial necrosis, intraluminal calcium deposits in the renal tubules, and glomerular fibrosis. He, like the brother who had had post mortem examination, had cerebral arteriosclerosis. Whether the mentally retarded mother had the genetic defect that led to convulsions and cardiovascular lesions in her sons, who might have been susceptible to earlier (fatal) manifestations of magnesium deficiency, having been born in rapid succession and thus probably with low stores of magnesium, is speculative. The infant who developed neonatal fits at eight days of life that did not respond to pyridoxine, glucose, or calcium therapy, but immediately improved following magnesium administration, had been born to a mother with celiac disease (Davis et al., 1965), and thus probably had low body stores of magnesium.
It is provocative that calcium, vitamin D, and sometimes PTH were used to control the neuromuscular irritability and to correct the hypocalcemia of almost all the infants and children ultimately found to be suffering from magnesium malabsorption. Their serum calcium generally rose, sometimes to hypercalcemic levels, but their clinical signs persisted (with lowered serum magnesium levels) until their magnesium deficiency was diagnosed and corrected. Infants with severe gastroenteritis or with PCM have also developed hypomagnesemia during the recovery period, while being fed diets rich in calcium, vitamin D, and protein.
Similarly, calcium therapy has not been effective in controlling postoperative seizures, or those developing after exchange transfusion, whereas magnesium therapy corrected the convulsions and both the hypocalcemia (Atwell, 1966; Dooling and Stern, 1967; Jalbert et al. 1969). Even feeding vitamin-D-fortified cows' milk to an infant recovering from a colostomy was found to produce hypomagnesemic (0.5 mEq/liter) convulsions that responded promptly to magnesium repletion (Savage and McAdam, 1967). Wilkinson and Harris (1969), who tested surgically treated infants for magnesium deficiency by the parenteral magnesium-load test (Thoren, 1963), found that there was severe depletion in 5 of 9 of their patients. In their further study, they found that 20 of 29 infants (many of whom had undergone gastrointestinal surgery) retained sufficient of the loading dose of magnesium to indicate deficiency, despite normal serum magnesium levels in four of nine whose serum levels were also measured.
Thus, the frequently spontaneous reported restoration of serum magnesium levels to normal, following moderate calcium treatment of infantile convulsions (David and Mast, 1974; D. R. Brownet al., 1976; Salle et al., 1977), is not absolute evidence that magnesium deficiency might not still be present. As had been indicated, there have been many instances of profound intensification of overt manifestations of infantile hypomagnesemic hypocalcemia by treatment with calcemic agents. In 1973, Volpe distinguished "jitteriness" from neonatal seizures, and commented that if hypocalcemic convulsions are refractory to calcium gluconate infusions, hypomagnesemia should be sought and treated by adding 2-3% magnesium sulfate (2-6 ml) to the intravenous infusion. He more recently (1977) commented that calcium infusions should not be given routinely to all newborns during their initial seizures, and recommended that if hypomagnesemia is present the magnesium should be given intramuscularly (0.2 ml/kg of 50% MgSO4 rather than intravenously. He noted that about half of newborns with seizures secondary to later-onset hypocalcemia also have hypomagnesemia, and that calcium administration to such infants may aggravate the hypomagnesemia and maintain the convulsive state.
It is not known whether the "jitteriness" of infants (such as is described in infants who died of the SIDS) is equivalent to the tremor syndrome reported from India as a manifestation of infantile magnesium deficiency (Wong and Teh, 1968; Chhaparwal et al., l971b, 1971/1973). Wong and Teh (1968) observed 13 of a series of 40 babies with convulsions or tremors of infancy who had hypomagnesemia in the absence of hypocalcemia. The remainder were low in both cations. Tremors, that developed on the first to third day of life (associated with serum magnesium levels of 0.66-1.14 mEq/liter) promptly responded to intramuscular 50% MgSO4 (0.5-1.5 ml/24 hours). A feeble infant, who had required resuscitation, and another whose tremors did not develop until the 30th day of life, required many injections to manage the recurrent tremors. These investigators also reported seven additional infants and young children with hypomagnesemic normocalcemic tremors responsive to magnesium therapy. They commented that the 13 babies with hypomagnesemia alone could not be clinically differentiated from 27 additional infants and young children who had hypocalcemia with and without hypomagnesemia. Radde et al. (1972), in their study of concomitantly low total magnesium and ionized calcium in infants with symptomatic hypocalcemia, also reported an occasional infant with convulsive hypomagnesemia alone. Cockburn et al. (1973) found only hypomagnesemia without hypocalcemia in 7% of their series of 75 convulsing newborn infants. In almost 80% there were combined mineral disturbances, low magnesium and calcium in half. "Jitteriness" was seen in 36% of those with hypomagnesemia and hypocalcemia. Forfar's group (Cockburn et al., 1973) commented that in the beginning of their study, before they realized the importance of hypomagnesemia in maintaining hypocalcemia and convulsions, they routinely gave calcium gluconate oral supplements to such infants. Calcium infusions were added if convulsions persisted. Later, treatment with 0.2 ml/kg 50% MgSO4 became routine. They found that giving intramuscular magnesium was more effective in raising the serum calcium than was oral calcium (Fig. 4-14A). With this treatment it became unnecessary to administer calcium intravenously. In fact, the found that magnesium alone restored both normal magnesium and calcium levels (Fig. 4-14B). They cautioned against overdosing with magnesium during the neonatal period, because of the risk of neuromuscular blockade, and allowed only two doses of magnesium per infant before redetermining serum levels. Four years later, this group analyzed the comparative results of treating neonatal tetany with magnesium sulfate alone, calcium alone, or a barbiturate (Turner et al., 1977). Among 10,500 live births over a 2 1/2- year period there were 104 infants with symptomatic hypocalcemia that started at 4 to 8 days of age. They were randomly allocated to three treatment groups: 34 were given calcium gluconate (10 ml of 10% solution orally with each feed for 48 hours); 33 were given phenobarbitone (7.5-15mg at 6-hour intervals); 37 were given 0.2 ml 50% MgSO4 intramuscularly. Mean posttreatment plasma calcium and magnesium levels were significantly higher in the magnesium-treated group than in either of the other groups, and the number of convulsions and number of treatments necessary to control the convulsions significantly lower (Table 4-1). Only one infant in the magnesium-treated group was still convulsing after 48 hours treatment, whereas 13 and 10 were still convulsing after 48 hours of calcium and barbiturate therapy, respectively (significance: p = 0.001). This group found the magnesium therapy to be free of major side effects, provided it is injected deep into the muscle, and recommend that magnesium sulfate is the treatment of choice for infantile hypocalcemic convulsions, whether or not hypomagnesemia is present. Paunier et al. (1974), who first detected the primary magnesium malabsorption syndrome (Paunier et al., 1965) has commented that the clinical syndrome of hypomagnesemia is indistinguishable from that of hypocalcemia. When the magnesium deficit is severe, as in the genetic disorder, he recommends intramuscular administration of 0.5-1 mEq of magnesium/kg body weight. He, too, cautions against intravenous administration because of the effect of hypermagnesemia on cardiac and neuromuscular conduction. Those with chronic hypomagnesemia are given 1-2 mEq/kg of oral magnesium salts in divided doses.
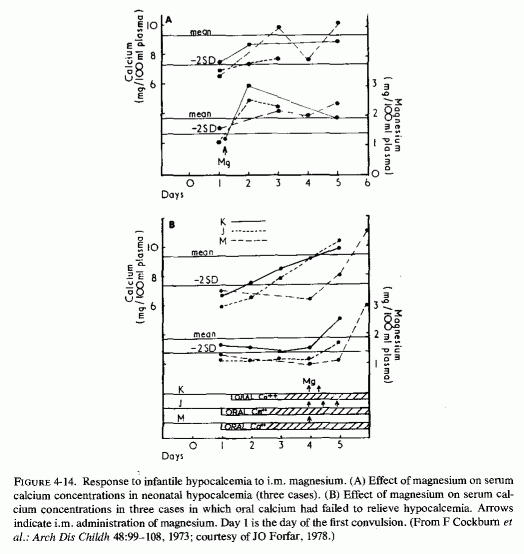{kind=link}
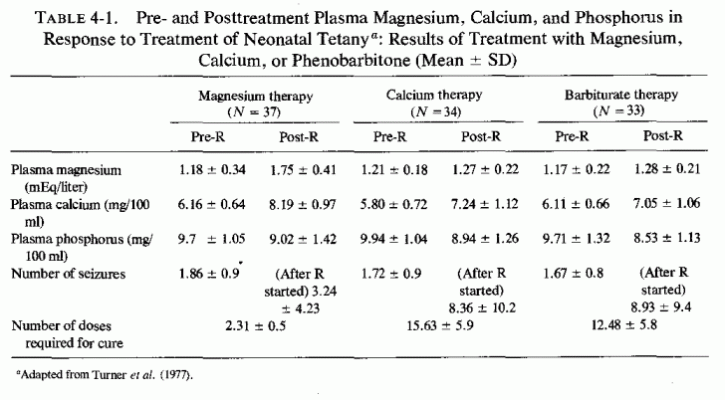{kind=link}
In view of the risk that not only convulsive disorders, which demand immediate attention, are a risk of calcemic rather than magnesium therapy, this author supports the conclusion of Forfar's group (Turner et al., 1977) that magnesium, not calcium, is the treatment of choice. Another caution must be given, applicable to infants and children whose hypocalcemia has been under treatment with such a calcemic agent as vitamin D. When magnesium is given to such patients, some respond to previously given vitamin D (which as a fat-soluble vitamin is stored) by developing sudden hypercalcemia. Durlach (1961), who observed that vitamin D therapy (in normocalcemic tetany) is effective only when the magnesium deficit is repaired, later cautioned that magnesium therapy restores the hypercalcemic response to high-dosage vitamin D, and that its administration should be carefully monitored by measurement of serum calcium when treating with magnesium (Durlach, 1969a, 1971). The observation that hypercalcemia has developed when magnesium therapy is added to high-dosage calcium and vitamin D therapy (i.e., of vitamin-D-resistant rickets: Rosier and Rabinowitz, 1973) suggests that release of PTH (Review: Anast, 1977), its conversion to an active form (Passer, 1976), or response to vitamin D might be subnormal in the presence of hypomagnesemia.
On the other hand, the classic treatment of vitamin-D-resistant osteopenias, which are usually associated with hypocalcemia, is with pharmacologic doses of calcemic agents. Vitamin D and its new metabolites are the most frequently used agents. It is well to recall that vitamin D poisoning is a risk, whether in the treatment of hypoparathyroidism (Leeson and Fourman, l966a,b) or in the treatment of vitamin-D-refractory rickets (Paunier et al., 1968a; Moncrieff and Chance, 1969). It is proposed that evaluation of the magnesium status, and a trial of magnesium therapy be given in vitamin-D-refractory rickets. It is conceivable that the magnesium might suppress the secondary hyperparathyroidism, thereby correcting the phosphaturia, and it might enhance both bone mineralization and formation of normal matrix.
The first reference found, with data on plasma magnesium as well as calcium levels in infants and young children, included 38 patients with magnesium determinations, 24 of which were low (Denis and Talbot, 1921). Half of those with hypomagnesemia (< 1.40 mEq/liter) were listed as having feeding problems (cited as "regulation of feeding"). Ten of those 12 had concomitant hypocalcemia (1.0-6.8 mg/l00 ml) and 1 had hypercalcemia (12.9 mg/100 ml). Most studies since then have stressed hypocalcemia as the predominant factor in neonatal tetany, a syndrome seen almost exclusively in bottle-fed infants. The higher phosphorus/calcium ratio of cows' milk, as compared to human milk, has been usually blamed. However, as the importance of hypomagnesemia has been recognized in many infants with hypocalcemic tetany, the high phosphorus/magnesium ratio of cows' milk has also been considered. The possibility of transient hypoparathyroidism and renal tubular immaturity has each been investigated as the explanation of the neonate's failure to correct the often long-sustained hyperphosphatemia that is derived principally from cows' milk. Forgotten is a provocative preliminary report (Swanson, 1932) that showed that an infant fed cows' milk from one to three months of age retained much more calcium than he did phosphorus or magnesium as compared with an infant of the same age fed human milk. When vitamin D (in cod liver oil) was added to the regimen of both infants at three months of age, their daily retention of all three elements rose. The differences in mineral retentions effected by the addition of vitamin D is mentioned here because the formulas administered in most of the subsequent comparative studies incorporated vitamin D; most infants receiving human milk were not so supplemented. Thus, the contrasting findings in breast-fed and formula-fed infants can be a consequence, not only of the higher mineral content and different phosphorus/mineral ratios, but a consequence of the difference in vitamin D supplementation. Not resolved is what happens to the excessive minerals retained by cows'-milk-fed infants. Manifestly, as indicated by the hypocalcemia and hypomagnesemia of artificially fed infants, the retained divalent cations must reach tissue sites, from which, probably as a result of hormonal imbalances, they are not readily mobilized.
4.3.1. Human versus Cows' Milk
The mineral content of human milk is considerably less than that of cows' milk (Table 4-2), the cows' milk being suited to the needs of the calf, which grows much more rapidly than does a human infant. The ratios of phosphorus to magnesium and calcium in the reconstituted dried cows' milk used in Scotland, and human milk, have been given by Cockburn et al. (1973) as follows:
{kind=link}
| Cows' milk | Human milk | |
| P/Mg | 7.5/1 | 1.9/1 |
| P/Ca | 0.8/1 | 0.2/1 |
The excessive phosphorus in cows' milk contributes to the abnormalities of serum levels of both calcium and magnesium, not only because of the higher dietary intake of phosphorus in formula-fed babies but because of functional factors (parathyroid and renal) that interfere with adequate elimination of the phosphate load and interfere with mobilization of bone minerals. The earlier studies stressed the phosphorus and calcium. The importance of magnesium in calcium homeostasis has been increasingly recognized, and more attention is now being paid to magnesium levels and to the influence of hypomagnesemia on hormonal function and calcium homeostasis.
Still largely disregarded is the role of the intake of vitamin D, despite the occasional comparative study of serum calcium, magnesium, and phosphorus levels in breast-fed versus bottle-fed infants that suggest the need for further work in this area.
The early long-term metabolic study (Swanson, 1932) performed on two infants 10-14 days to 6 months of age: one fed on pooled human milk except for a 1-week cows'-milk-consumption comparative period, and one fed cows' milk throughout, contains much thought-provoking data. This is the only study found in which the effect on mineral retention of whole cows' milk (without added vitamin D) was recorded. It also provides data on the change in mineral retention caused by addition of vitamin D (1 teaspoon cod liver oil) to the regimens of both infants, starting at three months of age(Table 4-3), although there were no signs of rickets. The ratios of mineral retention for the infant fed human milk to those for the cows'-milk fed infant were:
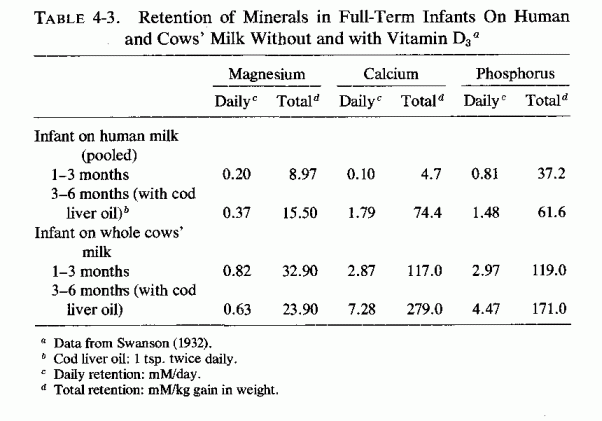{kind=link}
| Human-milk-fed/Cows'-milk-fed | ||
Months 1-3 | Months 3-6 (with cod liver oil) | |
| Mg | 1/3.6 | 1/1.2 |
| Ca | 1/25.0 | 1/3.8 |
| P | 1/3.1 | 1/3.1 |
The infant given human milk was switched to cows' milk for a 5-day metabolic period, before being continued on his usual regimen. During that period, his cumulative phosphorus retention increased twofold over each of the previous two 6-day metabolic periods; his cumulative calcium and magnesium retentions rose about fourfold over the average of the previous two periods. Shifting back to breast milk resulted in reversal of magnesium and calcium retentions to near prior values, but in a sharp (over tenfold) drop in phosphorus retention. Administration of cod liver oil to the infant on human milk initially resulted in a fall in retention of calcium, but there was a rapid increase thereafter, with an average daily retention in the last two metabolic periods more than tenfold greater than before the supplement was given. The average increases in daily phosphorus and magnesium retention were moderate, although phosphorus retentions rose much more in the last weeks of the study than in the first weeks after the vitamin D had been added (2-5 mM/6-day period to 12-15 mM/6-day period). Administration of cod liver oil to the infant on cows' milk increased his retention of calcium and phosphorus to lesser degrees, and decreased his magnesium retention.
The study reported by Slater (1961) compared mineral balances over observation periods of two to three days from the sixth to ninth days of life. They compared the balances in 13 breast-fed infants and 9 infants fed cows' milk formula (containing 317 IU vitamin D/400 ml reconstituted dried milk). The ratios of mineral retention for the breast-fed infants to bottle-fed infants were:
| Breast-fed/Bottle-fed | |
| Mg | 1/3-4 |
| Ca | 1/5 |
| P | 1/3 |
When additional phosphorus (120 mg/day) was given to the breast-fed infants, their urinary excretion of calcium dropped from the normal for breast-fed infants (4.43 ± 2.4 mg/kg/24 hr) to 2.07, close to that of bottle-fed babies (2.40). Their urinary phosphorus increased from 0.46 to 20 mg/kg/24 hr, but was still less than that put out by bottle-fed infants (34.9 mg/kg/24 hr). Their urinary magnesium dropped substantially from 0.61 to 0.19 mg/kg/24 hr (less than that on cows' milk: 34.9). The fecal output was not measured.
Despite the better retention of these minerals by infants on cows' milk as compared with that of breast-fed infants, it is among formula-fed infants that symptomatic hypocalcemia (often with hypomagnesemia) constitutes a problem. Thus, subsequent studies have been done with cows' milk adapted to resemble mothers' milk more closely. Widdowson (1965) compared mineral retentions by infants fed human and adapted cows' milk (Table 4-4). She observed several striking differences in retentions. Most notable was the low calcium retention during the fifth to seventh days of life in the formula-fed infants, as compared with that of breast-fed infants. By the fourth to seventh weeks, the calcium retention was greater in infants on one of the formulas and less in those on the formula that, paradoxically, delivered the greatest amount of calcium, than it was in the breast-fed infants. The phosphorus retentions were greater in all of the formula-fed infants than in the breast-fed infants, and the magnesium retentions of the formula-fed infants were the same or greater than those of the breast-fed infants. This study confirmed, by showing the poor retention of calcium by the young neonate on cows' milk, the greater susceptibility of infants fed cows' milk than breast fed infants to calcium insufficiency. The high content of phosphorus and saturated fats of cows' milk has each been implicated in the hypocalcemia Oppé and Redstone, 1968; Widdowson, 1969; Barltrop and Oppé, 1970; Pierson and Crawford, 1972) but each of these factors would also cause interference with retention of magnesium.
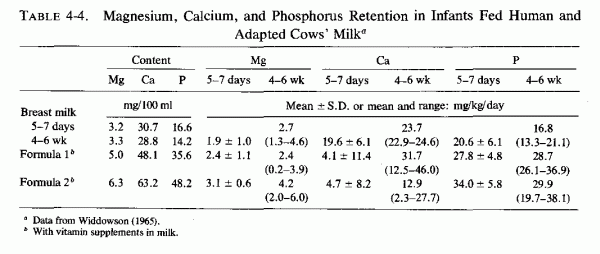{kind=link}
Hyperphosphatemia, and a wider than normal range of serum calcium levels are frequently encountered in normal infants fed cows' milk formulas from birth, abnormalities that are in contrast to ranges within normal limits in most normal breast-fed infants. Studies of comparative serum calcium and phosphorus values in normal infants were undertaken when it was found that infants with neonatal tetany had hypocalcemia and hyperphosphatemia and that this syndrome was virtually unknown where breast-feeding was customary. Bakwin (1937) considered the high phosphorus content of cows' milk to be contributory to persistent neonatal hyperphosphatemia, which he believed might be intensified by transient hypoparathyroidism, such as had been proposed by Pincus and Gittleman (1936) to explain nonrachitic tetany in a 7-week-old infant. They found that feeding infants phosphate solutions resulted in just such a rise in serum phosphorus and fall in serum calcium as is seen in neonatal tetany. Immaturity of the kidneys, with inability to clear phosphate at normal (adult) rates, was proposed by Dean and McCance (1948). Both theories have been substantiated, although new insights have recently been acquired.
L. Gardner et al. (1950) studied 16 cases of tetany that provided support for the etiologic role of the high P/Ca ratio of cows' milk (which all 16 infants with tetany had been fed). They also showed that the maximum renal P clearance of the infants was only 10% of the probable glomerular filtration rate [shown to be less than half that of adults (Dean and McCance, 1947)]. This they attributed to prenatal factors, such as maternal hyperparathyroidism with secondary neonatal hypoparathyroidism. They also considered serum magnesium levels in normal infants on different feedings, in an attempt to elucidate the cause of neonatal tetany, and showed that even normal newborn infants on formula had pronounced falls in total serum magnesium that were accompanied by decreased ionized calcium and increased serum phosphorus levels. A premature infant shifted from human to cows' milk promptly exhibited a rise in serum P from 6.45 to 11.26 mg/100 ml, that dropped to the original level several days after reinstituting human milk feeding.
The studies of Oppé et al. considered only the serum calcium and phosphorus levels of bottle-fed and breast-fed infants and confirmed that the latter had significantly lower serum phosphorus and higher serum calcium levels than the former (Oppé and Redstone, 1968). Infants fed cows' milk adapted to resemble breast milk had the same mean serum calcium levels as did breast-fed infants although there were more with hypercalcemia and several with marginal hypocalcemia, not seen in the infants on breast milk (Fig. 4-15A). The lowest range of serum phosphorus levels was in the breast-fed infants; that in adapted cows' milk was lower than in unadapted cows' milk, but higher than levels in breast-fed infants (Fig. 4-15B). These investigators commented that early addition of cereals (with their high phosphorus as phytate content) to the infants' diets can increase the tendency toward hypocalcemia. It should be noted that phytates also interfere with absorption of magnesium. Two years later, this group published its further studies of the factor(s) in cows' milk responsible for the induction of infantile hypocalcemia, resulting in the symptomatic neonatal tetany that is seen, usually by the fifth to seventh days of life of formula-fed normal-birth-weight infants (Bar and Oppé, 1970). They used milk preparations with altered calcium and phosphorus contents, and found that neither is solely responsible for the hypocalcemia. They considered the ratio of dietary Ca/P most important. Addition of calcium to cows' milk formula fed to low- birth-weight infants increased their calcium retention (Barltrop and Oppé, 1973). Feeding low-birth-weight infants (4-41 days of age) formulas differing in calcium and phosphate contents exerted little influence on the plasma calcium and phosphorus levels, which varied widely (Bar et al. 1977). The investigators commented that additional factors (than calcium, phosphorus, and fat contents of the formula) require study. They did not explore the magnesium levels; all of the cows' milk formulas used incorporated vitamin supplements (Widdowson, 1965).
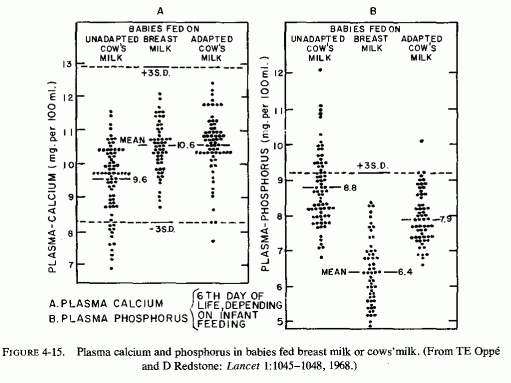{kind=link}
The effect of vitamin D on the serum calcium and phosphorus levels of infants fed cows' milk or breast milk was studied by Pincus et al. (1954). They analyzed levels on the day after birth and on the fifth day of life (Figs. 4-16A, B). All of the infants on cows' milk had significantly higher serum phosphorus levels on day 5 than did the breast-fed infants, whether or not they were given vitamin D. They observed that administration of vitamin D to formula-fed infants, in the first five days of life, increased the incidence of hypocalcemia (below 8 mg/100 ml) from 10.9% in infants without vitamin D to 17.3% of those who were given vitamin-D fortified milk (400 USP units/quart), and to 30% of those given nonfortified milk, but a higher dose of vitamin D (600 units daily in an aqueous preparation of multi-vitamins). This finding is in accord with the later observation that 5- to 7-day-old infants on cows' milk retained little calcium (4.1-4.7 mg/kg/day) as compared with that of breast-fed 5-to 7-day-old infants (19.6 mg/kg/day) who were given no vitamin supplements (Widdowson, 1965). Breast-fed infants, given the same vitamin preparation, exhibited no such change in calcium levels (Pincus et al. ., 1954). This group later showed that vitamin D also played a role in neonatal hypomagnesemia of formula-fed infants (Gittleman et al. 1964). They found that the serum magnesium levels of neonatal infants dropped minimally after five days of cows' milk formula, without vitamin D added, in contrast to the slight rise in serum magnesium of breast-fed infants. Administration of 600 units of vitamin D resulted in lower serum magnesium levels (from means of 1.75 to 1.5 mEq/liter on day 5) in the bottle-fed infants, but no change in infants on mothers' milk. Serum phosphorus levels rose by day 5 in bottle-fed infants, with our without vitamin D, but did not rise in any of the breast-fed babies.
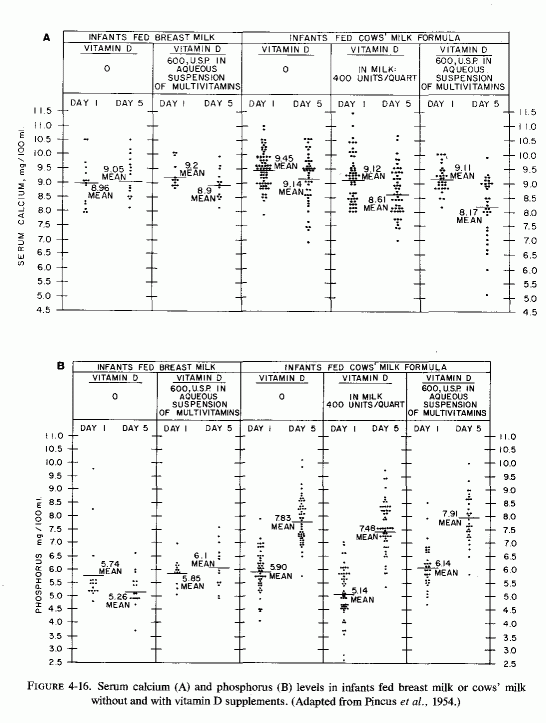{kind=link}
In the study of normal neonatal infants by Gardner et al. (1950) that showed increased serum phosphorus and decreased total magnesium and ionized calcium in those that were on formula, each bottle-fed newborn infant was given 750 units of vitamin D3 whereas the breast-fed infants received no vitamin supplements.
Anast (1964) studied serum magnesium levels in a large group (72) of normal full-term infants who were born without complications after normal pregnancies. Almost half (34) were breast-fed and received no vitamins; the remainder (38) were given evaporated milk formulas containing 400 units of vitamin D. He found the mean serum magnesium levels of bottle-fed babies to be lower than that of breast- fed babies on days 3-5, and attributed the difference to the high phosphorus content of cows' milk. In a smaller study (22 formula-fed infants and 5 breast-fed infants) no difference was found in serum magnesium values (Bajpai et al., 1966).
In contrast, Ferlazzo et al. (1965) found that breast-fed infants had slightly lower serum magnesium levels (1.5 mEq/liter) than did infants given half-cream cows' milk (1.7 mEq/liter). They speculated that this difference might reflect maternal hypomagnesemia.
Plasma calcium, magnesium, and phosphorus levels of bottle-fed and breast- fed infants were compared by Harvey et al. (1970). Among normal formula-fed infants, the mean plasma phosphate level was 8.25 mg/100 ml, with levels reaching as high as 21, as compared to a mean of 6.25 in breast-fed infants, none of whom had plasma P above 9.8 mg/100 ml. The plasma magnesium levels were significantly lower (p < 0.001) on the sixth day of life in the bottle-fed infants than in breast-fed infants. At that time the mean levels of magnesium were 0.91 mEq/liter and 1.33 mEq/liter, respectively, and the mean levels of calcium were 7.6 and 8.6 mg/100 ml for normal bottle-fed and breast-fed babies. The ranges of levels were wider in bottle- than breast-fed infants.
| Bottle-fed | Breast-fed | |||
| Plasma Mg (mEq/liter) | 0.67-1.6 | 1.0-1.7 | ||
| Plasma Ca (mg/l00 ml) | 3.8-11.2 | 8.0-12.4 | ||
| Plasma P (mg/l00 ml) | 4.6-21.0 | 4.1-9.8 |
Convulsing infants in this study had mean plasma magnesium levels lower than did breast-fed infants, but equal to levels of bottle-fed infants (0.9 mEq/liter). Their mean serum calcium level (6.3 mg 100 ml), however, was lower than that of bottle- fed normal infants (7.6 mg/100 ml). Snodgrass et al.(1973) also observed a greater rise in serum magnesium and calcium levels from the first day of life to days 6-8 in breast-fed versus formula-fed infants
Forfar et al. (1971/1973) reported that normal, breast-fed infants had serum magnesium levels on the sixth day of life that equaled that in cord blood, whereas those on cows' milk showed a decline in serum magnesium levels during the second to sixth days. Convulsions of infancy that occurred from the fourth day onward in 62% of the infants, were associated with plasma magnesium concentrations below the normal range in 65% of the cases. There was a strong positive correlation between magnesium and calcium levels (p < 0.001) and a lesser but still significant negative correlation between magnesium and phosphorus levels (p < 0.01). In a study of 75 additional consecutive newborn infants with convulsions, these investigators observed that all of the convulsing infants were fed an evaporated milk formula (Cockburn et al., 1973). Figure 4-3 depicts the comparative values for plasma calcium, magnesium, and phosphorus concentrations for (normal) breast-fed infants and for the infants with convulsions. They also found that both mean plasma values and ranges for these elements differed significantly in breast-milk and normal cows'-milk-fed infants, particularly on the fifth to seventh days of life. They considered the possibilities, suggested in the literature, that the high phosphorus load provided by cows' milk might exert a hypocalcemic effect mediated by transient hypoparathyroidism (Fanconi and Prader, 1967), maternal calcium or vitamin D deficiency (Watneyet al., 1971) or either vitamin D administration (Gittleman et al, 1964), or deficiency (Barr and Forfar, 1969) in the infant. They noted that infantile hypomagnesemia similarly might result from the disproportionate phosphorus load, mediated by transient hypoparathyroidism. The better response to magnesium than to calcium therapy of neonatal tetany, and the risk of aggravating the hypomagnesemic convulsive state was also noted. This may constitute reconsideration of an earlier recommendation that the hypercalcemic agent, vitamin D, be given in high doses (5000 IU/day) in the treatment of hyperphosphatemic (hypocalcemic) tetany of the newborn (Barr and Forfar, 1969). It is noteworthy that comments have been made in textbooks that vitamin D is ineffective in transient neonatal tetany (Nelson, 1964) and might be dangerous (Fourman and Royer, 1968).
That accepted prophylactic doses of vitamin D can lower the serum magnesium levels of normal infants, though only slightly, is an observation that should be considered in light of the findings that (1) vitamin D excess causes magnesium loss; (2) there is a broad spectrum of reactivity to vitamin D(Fig. 4-17, Fanconi, 1956) (Reviews: Seelig, 1969b, 1970a,b), and that fortification of milk and other foods makes intakes of higher than prophylactic amounts almost unavoidable. It is possible that such high intakes of vitamin D in infancy, when phosphate intakes are also likely to be high (in bottle-fed babies) and there is risk of magnesium deficiency, can contribute not only to the acute infantile manifestations of abnormal calcium and magnesium homeostasis, but to early and later cardiovascular skeletal, and renal, diseases (Seelig and Haddy, 1976/1980).
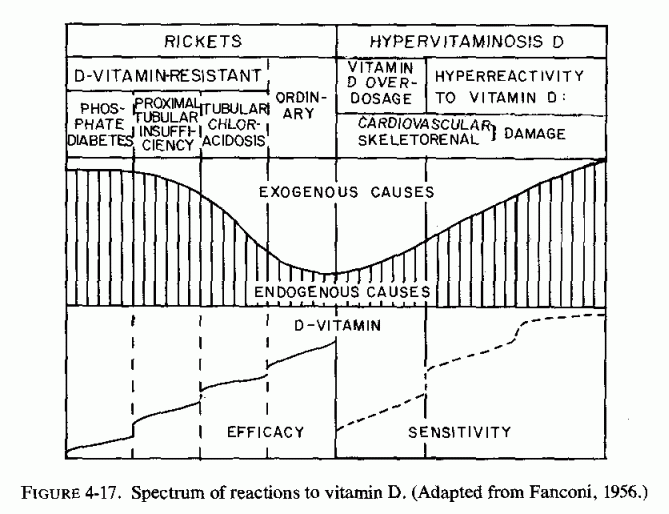{kind=link}
There is wide variation in susceptibility to vitamin D toxicity and in the requirements for vitamin D, both in experimental animals and in man (review: Seelig, 1969b). Thus, whether infants will develop early or late sequellae of hypervitaminosis D depends upon their tolerance of the therapeutic amounts of vitamin D most ingest for prophylaxis of rickets. The Food and Nutrition Board of the American National Research Council (1968 Edition) commented that normal full-term infants require as little as 100 IU of vitamin D daily to prevent rickets and that premature infants usually require no more than 200 IU. As a result of the outbreak in Great Britain of infantile hypercalcemia, with resultant supravalvular aortic stenosis syndrome (SASS), comprising arterial as well as valvular lesions, renal damage, growth failure, a typical peculiar facies, and severe mental retardation which resembles that reported by Williams et al. (1961) (Reviews: Black, 1964; Seelig, 1969b) (Fig. 4-18), there was intensive reevaluation of the possible causative role of hypervitaminosis D. A Committee of the British Paediatric Association reported that the intakes of vitamin D by British infants might well reach 4,000 IU daily if all of the available fortified infant foods and supplements were consumed (Committee Report, 1956)-this despite their earlier (1943) cited recommendation that the total daily intake of vitamin D should not exceed 500-700 units. There was also an outbreak of the SASS and other outflow obstructions (Fig. 4-19) in Germany where extremely high doses (Stosstherapie: 200,000-300,000 units) were injected two or three times a year (Beuren et al., 1964, 1966). Even in the United States, where the American Medical Association Council on Food and Nutrition had refused to countenance more than 400 IU vitamin D per quart of milk (F. Bing, 1941), many cases have been reported (Seelig, 1969b) (Figs. 4-20, 4-21, 4-22) that are indistinguishable from those associated with moderate to extreme overdosage with vitamin D. Classic SASS, originally described only in fair-skinned children (Williams et al., 1961; Seelig, 1969b), has also been reported in black children (Kostis and Moghadem, 1970; Fig. 4-22). Cardiac outflow abnormalities, whether as the entire complex of SASS, as an incomplete picture (Fig. 4-21) with stenosis of the right outflow of the heart with or without notable mental retardation and/or cardiofacies (Figs. 4-23, 4-24, 4-25), or as part of a more generalized picture of "congenital" cardiovascular disease is now so prevalent that the literature is replete with papers describing individual or familial instances, diagnostic procedures, and techniques for surgical repair. This complex of diseases had been so rare before the l930s as to have been omitted or given only passing reference in most textbooks and atlases of cardiovascular pathology (Perou, 1961). Congenital disorders associated with an exogenous etiologic factor (as the thalidomide-induced teratology), are characterized by a wide range of malformations, depending on the magnitude, time, and extent of the insult (Taussig, 1965, 1966; Beuren et al., 1966). Thus, one should anticipate a similar variety of abnormalities associated with the damage caused by the nutritional imbalances that are part of the hypervitaminosis-D-complex (Taussig, 1965, 1966). That such a variety is likely to exist is indicated by the different findings reported in victims of hypervitaminosis D and in relatives. Multiple arterial stenoses were described in infants who died early with severe infantile hypercalcemia (Bonham-Carter and Sutcliffe, 1964), and coexisting bilateral pulmonary artery stenosis, as well as additional cardiovascular abnormalities, depending on the degree and time of vitamin D overdosage. The mental and facial abnormalities were not consistent. In a particularly interesting family with 11 cases, nine of whom had supravalvular aortic stenosis without cardiofacial appearance and mental retardation, two died in infancy with generally hypoplastic major arteries before the aortic stenosis had developed. One died at seven months after unsuccessful attempts to control his infantile tetany with dihydrotachysterol (0.6 mg/day) and vitamin D (1,000 IU/day) had failed, despite hypercalcemic response shortly before death. His cousin died suddenly at three weeks of age, a week after a vitamin D treatment (Beuren et al., 1966). This paper dealt with 54 patients, in most of whom there was a clear history of "Stosstherapie." Several of the mothers also admitted to continuous vitamin D supplementation during pregnancy. Occurrence in one family of instances of sudden infant death, hypercalcemia, hyperreactivity to vitamin D, and a wide range of cardiovascular stenotic and hypoplastic pathologic changes, with and without peculiar facies and mental retardation, suggests a common pathogenesis. In the family reported by Beuren et al.(1966) and in isolated unrelated and other familial cases, there was strong circumstantial evidence that those who developed the syndrome were unable to detoxify the excessive parenteral doses of vitamin D that was a common mode of prophylaxis against rickets in Germany at that time.
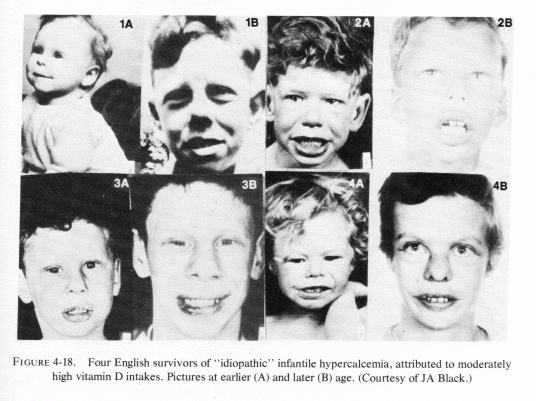{kind=link}
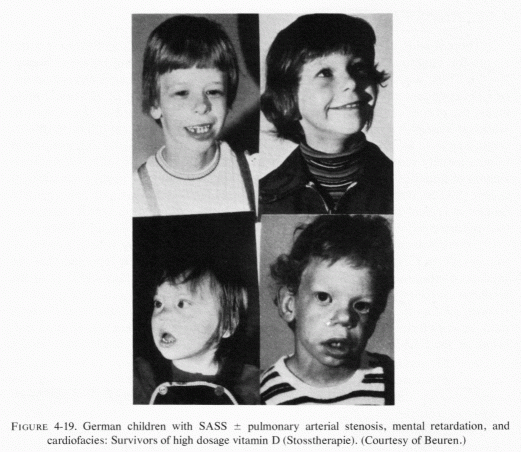{kind=link}
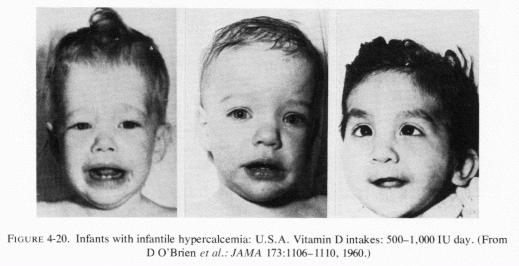{kind=link}
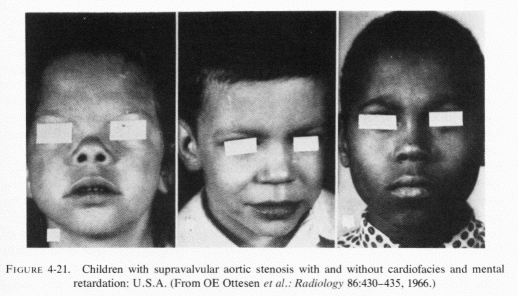{kind=link}
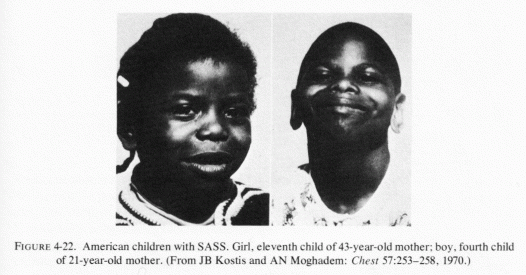{kind=link}
{kind=link}
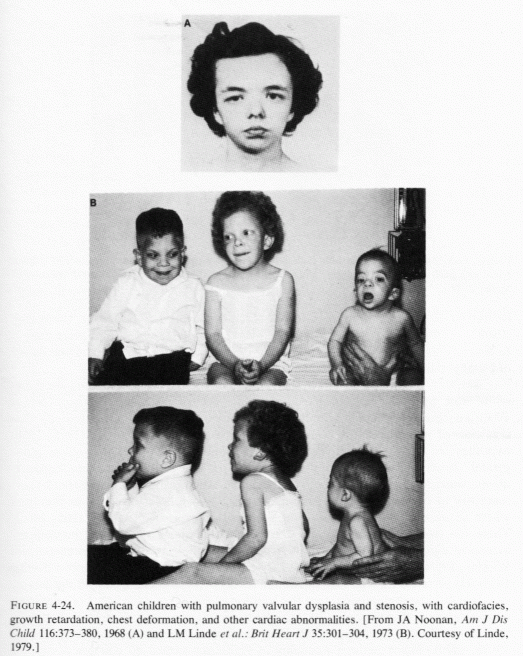{kind=link}
{kind=link}
The similarity to the SASS of the syndrome, seen in England among survivors of infantile hypercalcemia (Schlesinger et al., 1956, Black and Bonham-Carter, 1963), suggested that some infants were so susceptible to vitamin D toxicity that ingestion of lesser amounts could cause permanent injury. Taussig (1965, 1966) hypothesized that hyperreactivity to vitamin D might well be the cause of the "congenital" heart disease: SAS, and of gradations of injury. Because hypercholesterolemia was found in some of the hypercalcemic infants, she speculated that hyperreactivity to vitamin D might be contributory to hypercholesterolemia in countries where vitamin D supplementation of foods is widespread (Taussig, 1965, 1966). There has been experimental and epidemiologic evidence that even moderately increased vitamin D intakes have increased blood cholesterol levels (Feenstra and Wilkins, 1965; Dalderup et al., 1965: Linden, 1974b, 1975/1977; Linden and Seelig, 1975). Hypertension is also seen in vitamin D toxicity and in children with the SASS.
It should be remembered that the addition of 400 IU to each quart of milk is an amount arrived at empirically, because that amount of vitamin D delivered in milk was more effective in curing rickets than the same amount in oil (Reviews: Seelig, 1969b, 1970). The American Academy of Pediatrics expressed concern about the total vitamin D consumption in the United States, which they calculated might range from 600 to 4,000 IU daily from marketed fortified products (Committee Report, 1963). They recommended that no more than 400 IU should be provided from all sources, including sunlight, and reiterated and amplified their concern about hypervitaminosis D two years later, stressing the possible role of maternal factors (Committee Report, 1965). In consultation with the Committee, D. Fraser (1967) wrote a report reaffirming the limitation of vitamin D to no more than 400 IUday, and referred to evidence that as little as 100 IU or less has protected against rickets (Drake, 1937; Glaser et al., 1949). Despite these official recommendations, fortification of many foods with vitamin D persists, and many Americans supplement their diets with vitamin-D-containing vitamin preparations. Studies of dietary intakes show that, both in Canada and the United States, vitamin D intakes are often excessive (Dale and Lowenberg, 1967; Broadfoot et al., 1972). A Canadian study of 1,000 children one week to five and a half years of age showed that 70% ingested more than 400 IU daily and 31% more than 1,000 IU daily (Broadfoot et al., 1972). The narrow toxic/therapeutic ratio for vitamin D in infants (Stewart et al., 1964), and the wide differences in the amounts of vitamin D that are required or can be tolerated support D. Fraser's (1967) call for reappraisal of national policies concerning vitamin D requirements. He referred to the known toxicity of vitamin D and to the lack of knowledge concerning possible long-term effects of intakes from infancy that exceed requirements severalfold.
It is possible that the increased incidence, since the 1930s, of children's diseases that used to be rare and that have characteristics that resemble those seen in vitamin D toxicity might be consequences of the concomitant widespread and sometimes intensive use of vitamin D. The profound changes in the pediatric picture, in the twenty-odd-year period from early in the 1930s to 1965, led Hutchison (1955) to raise the point ". . . it is just possible that the very measures which we have used to abolish rickets from the land may have resulted in the appearance of hypercalcemia in some susceptible infants." The new diseases he cited were infantile hypercalcemia, infantile renal tubular acidosis and fibrocystic disease of the pancreas, usually with marasmus and steatorrhea, and cystinosis. The first two of these disorders have been definitely correlated with overdosage or hyperreactivity to vitamin D (Fig. 4-26, Lightwood and Butler, 1963; Review: Seelig, 1969b). Renal sclerosis and skull and other bone deformities are common in victims of SASS (Seelig, 1969b), and skeletal abnormalities are also seen in other outflow obstructive disease, such as of pulmonary stenosis (Noonan, 1968; Linde et al.1973). It is of interest that congenital valve disease is not uncommon in osteogenesis imperfecta. It has been suggested that mucoviscidosis might also be a consequence of hypervitaminosis D (Coleman, 1965). To what extent magnesium loss caused by excess vitamin D might contribute to sequellae of infantile hypercalcemia is not certain.
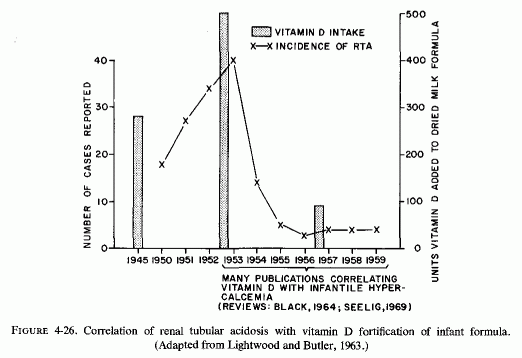{kind=link}
Moncrieff and Chance (1969) have pointed out that the margin between the therapeutic and the toxic dose of vitamin D is narrow, and described small calcium deposits in renal biopsy specimens of four children with hyphosphatemic rickets. Hypophosphatemic rickets, which had initially been treated with massive doses of vitamin D, was found to be associated with hypomagnesemia (0.5 and 0.7 mEq/ liter) in a five-year-old boy and a two-year-old girl (Reddy and Sivakumar, 1974). Despite concomitant hypocalcemia, there had been no convulsions. Magnesium therapy caused correction of the mineral abnormalities in the serum. It is possible that the latter two young children also had early renal damage, such as Moncrieff and Chance (1969) described, that resulted in renal wasting of magnesium. It has also been postulated that, since conversion of vitamin D to its active metabolites involves magnesium-dependent enzymatic steps, vitamin-D-resistant rickets might be a consequence of decreased formation of the active metabolites as a result of magnesium deficiency (Rosier and Rabinowitz, 1973). In the 13-year-old girl, whose magnesium-responsive vitamin-D-resistant rickets was hyperphosphatemic, PTH administration produced phosphaturia, but did not correct her symptomatic hypocalcemia until her hypomagnesemia (0.5 mEq/liter) was treated (Rosier and Rabinowitz, 1973). Thus, in this child with idiopathic hypoparathyroidism, magnesium depletion might have been primary, and causative of impaired bone response to PTH. Whether the children with hypophosphatemic vitamin-D-resistant rickets reflect an overt hyperparathyroidism secondary to hypomagnesemia is a possibility that deserves consideration. If the abnormal response to vitamin D is secondary to magnesium deficiency, attempting to treat the condition by this agent, which increases magnesium loss, can be responsible for damage that may not be manifest immediately. Since magnesium deficiency has been shown to cause osteoporosis in experimental animals, the osteopenia of vitamin-D-resistant rickets might be mediated in part by magnesium deficiency. The excess vitamin D given in the face of the magnesium deficiency, which in itself causes renal and cardiovascular damage, can intensify those lesions.
The cardiovascular lesions (that are related to the SASS) that cause death in earlier infancy from coronary or generalized arteriosclerosis, or that might be the pediatric precursors of adult atherosclerosis, might well be the result of nutritional and hormonal imbalances, to which vitamin D excess contributes. It is of interest to note that it was at the beginning of the era referred to by Hutchison (1955) as being marked by the emergence of new pediatric diseases, that Lightwood (1932) suspected hyperreactivity to vitamin D as a possible etiologic factor in the first published case of what was probably a late form of severe infantile hypercalcemia. He described a retarded, dwarfed two-year old girl who died with widespread endarteritis obliterans; endocardial calcification, hypertension, calcerous renal tubular casts, and osteosclerosis. In 1956, as chairman of a committee of the British Pediatric Association assigned to investigate the relationship of vitamin D (added to milk and other infant foods) to the virtual epidemic of infantile hypercalcemia, he recommended that the amount of vitamin D given to infants be sharply reduced, the maximum amount permitted, from all sources, to be determined after further investigation. That investigation is yet to be undertaken.
The first infant, whose primary hypomagnesemia and secondary hypocalcemia was associated with convulsions that were responsive only to magnesium, was found to have isolated intestinal malabsorption of magnesium (Paunier et al. 1965, 1968b). Even with about five times the normal oral intake of magnesium, this boy's serum magnesium levels remained at 1.1-1.4 mEq/liter. On discontinuing the supplement for a few days at 10, 18, and 30 months of age, there were further decreases in serum magnesium levels. PTH administration caused hypercalcemia without affecting the serum magnesium; large doses of vitamin D also increased the calcium level, but caused a gradual fall in serum magnesium, even when the infant received magnesium supplements. Magnesium malabsorption has persisted throughout the eight years of observation; interrelationships of his chronic magnesium deficiency with PTH were then evaluated (Suh et al., 1973).
The second patient with this abnormality was reported by Salet et al., (1966). They identified isolated malabsorption of magnesium, but normal renal magnesium conservation. They considered the condition congenital and later reported it to be a familial cause of primary hypomagnesemia, when a new sibling was found to have the same disorder (Salet et al., 1970). Like the first infant (Paunier et al., 1965, l968b), this child responded to exogenous PTH by increased serum calcium but not change in serum magnesium (Salet et al., 1966). Initially, his urinary output of phosphorus had been very low, and he had hyperphosphatemia. High-dosage vitamin D caused hypercalcemia and increased magnesium requirements. Magnesium therapy corrected the hypocalcemia and hypomagnesemia, and lowered the serum phosphorus level.
Infantile hypomagnesemia, as a result of malabsorption of magnesium (but normal renal conservation of magnesium) was reported in a boy born to first cousins, suggesting that this might be an hereditary disease with recessive genetic characteristics (M. Friedman et al., 1967). This infant's convulsions began on the 23rd day of life, were intensified by calcium therapy and subsided, as did his irritability and twitching, following parenteral magnesium therapy. Metabolic balance studies showed that he required over 600 mg of magnesium daily to sustain positive magnesium and calcium balances.
The cited cases were in French-Canadian (Paunier et al 1965, 1968b), French (Salet et al. 1966, 1970), and Indian (M. Friedman et al. 1967) male infants. A Norwegian male infant with comparable manifestations was reported by Skyberg et al. (1967, 1968). As in the other cases, despite large-dosage oral magnesium supplementation, the infant's serum magnesium levels remained subnormal, 1.3-1.4 mEq/liter, and fell further on temporary discontinuation of the supplements. PTH exerted no effect on the low serum magnesium, but raised serum calcium and increased phosphaturia twofold, during a short period in which magnesium supplements were withheld. Identification of the same disorder in two Norwegian brothers by the same group of investigators (Stromme et al., 1969) led them to term the condition "familial hypomagnesemia." The first of the brothers had died at 50 days of life, with continuous seizures associated with hypocalcemia that had been unresponsive to intravenous calcium or to vitamin D or anticonvulsive therapy. No magnesium determinations had been performed. When the second brother developed convulsions the third week of life, hypomagnesemia was identified. His hypocalcemia and slight hyperphosphatemia, as well as his seizures, subsided in response to intravenous magnesium administration. His serum magnesium remained subnormal (1.1- 1.4 mEq/liter) while receiving high-dosage oral magnesium supplementation. On its temporary discontinuation, he again gradually developed severe hypomagnesemia.
A Swedish female infant, who developed convulsions at two months of age, had hypomagnesemia, hypocalcemia, peripheral edema, and bulging fontanelles on admission (Haijamae and MacDowall, 1972). She required continuous high dosage oral magnesium supplements. After withdrawal of the magnesium supplements, her serum magnesium dropped from 1.3 to 0.5 mEq/liter, and her serum phosphorus rose from 4.8 to 6.0 mg/100 ml. Serum calcium rose and serum potassium fell slightly. More significant were the skeletal muscle electrolyte changes: Magnesium and potassium levels fell 9.5% and 7% respectively; muscle sodium rose by 53%.
Nordio et al. (1971) have intensively studied an Italian boy, who was hospitalized at seven months of age with convulsions and tetany that had not responded to oral calcium and vitamin D therapy. When he failed to improve following intravenous calcium, his plasma magnesium was measured and found to be 0.67 mEq/liter (normal range: 1.7-2.1). Hypoparathyroidism and possible magnesium deficiency were deemed likely, and he was given PTH and magnesium (2 g) intramuscularly, with partial improvement. He required high oral intakes of magnesium (30-70 mg/ kg/day) to normalize his clinical picture, and to correct the abnormal electroencephalogram, electromyogram, and electrocardiogram as well as serum calcium and phosphorus. His plasma magnesium did not attain normal levels. Each time magnesium supplementation was stopped, there was recurrence of irritability and tetany. His tissue potassium/sodium ratio was found to be low. He had higher than normal sweat concentrations of magnesium, but normal erythrocyte and cerebrospinal fluid levels of magnesium. He was proved to have selective intestinal magnesium malabsorption; his kidneys were able to conserve magnesium when he had hypomagnesemia. His intestinal mucosal ATPase seemed normal when tested in a medium containing MgSO4 Electron microscopic examination of his intestinal mucosal cells showed dilated endoplasmic reticulum and mitochondrial swelling in the apical portion of the cells. The brush border was normal. Long-term oral magnesium therapy prevented recurrence of the hypocalcemia, but his serum magnesium remained below the normal range, although not at the severely hypomagnesemic level found when he was seven months old. He was mentally retarded (I .Q. at 32 months was 68).
Woodard et al. (1972) reported diarrhea and anasarca as prominent manifestations that remitted with magnesium therapy in an American infant boy (two months old) whom they found to have selective magnesium malabsorption. Before detection of severe hypomagnesemia (0.06-0.1 mEq/liter), he had received calcium and anticonvulsant therapy for his generalized seizures, without improvement of either his hypocalcemia or his convulsions. Seizures abated after starting i.m. MgSO4 therapy, and soon the diarrhea and edema cleared. To avoid recurrence of his seizures, the infant required more than 200mg Mg daily, by mouth. His serum calcium had failed to rise in response to PTH while he was magnesium depleted; he developed a hypercalcemic response to its injection following magnesium repletion.
A Belgian boy, the ninth child of a mentally defective mother, was the fifth male sibling to have had convulsive attacks. Two brothers had died in the second and third months of life, respectively, having had generalized seizures; another had a single convulsion at six years of age, and a fourth had a seizure at 13 months (Vainsel et al., 1970). The child, whose magnesium deficit was identified shortly before his death, had been hospitalized with convulsions, peripheral edema, and bulging fontanelles. He had constant tetany, bilateral Trousseau sign, and carpopedal spasm. He seemed unaware of his surroundings and, except for intensification in response to noise, did not respond to stimuli. He was given intravenous calcium on detection of hypocalcemia, which raised his serum calcium level from 6.15 to 8.5 mg ml without improving the tetany. He had increased serum alkaline phosphatase, but normal phosphate levels. He was given high-dosage therapy of vitamin D (750,000 IU/week), which was stopped when hypomagnesemia was reported (0.4-0.65 mEq/liter). Parenteral therapy with magnesium was started, which raised his serum Mg to 2.3 mEq/liter. His tetany persisted until his death on the third day of the treatment with magnesium. Postmortem examination disclosed focal myocardial necrosis, calcinosis around a branch of a cerebral artery, and intimal calcification in another. Intraluminal calcium deposits were found in the proximal renal tubules and in the ascending limb of the loop of Henle. There was fibrosis and basement membrane proliferation in some glomeruli. He also had meningeal thickening and infiltration, a finding that had been reported in one of his brothers who had had seizures, and cerebral intimal calcification. A presumptive diagnosis of familial magnesium-malabsorption was made, on the basis of the similarity of the findings to those that had been described in the literature.
In retrospect, it seems likely that the boy (white American) with a history of similar manifestations-repeated convulsions, cyanotic attacks, tremors and nervousness from six months of age, for which he had been maintained on oral calcium and vitamin D supplement-might also have been a child with primary magnesium deficiency (J. F. Miller, 1944). He developed osteochondritis at 3½ years of age. When he was hospitalized (for malaria) at 6 years of age, and developed severe muscle cramps as well as carpopedal spasm and Trousseau sign, in the absence of hypocalcemia, he was found to have low serum magnesium (1.4 mEq/liter). All of his neuromuscular irritability subsided on oral magnesium supplementation; it recurred when treatment was stopped for a week. Marked hypomagnesemia, hypercalcemia, and hypophosphatemia were then observed, and again there was favorable response to oral magnesium therapy. Stromme et al. (1969) pointed out that this boy's early manifestations were similar to those of familial hypomagnesemia. Another boy with osteochondrosis, who has renal magnesium wasting (Klingberg, 1970), has developed myocardiopathy and peripheral muscle weakness (Klingberg, personal communication), all of which fit the general picture of magnesium depletion. Whether his initial lesion might have been magnesium malabsorption, as seems probable in a patient reported in Vainsel et al., (1970), cannot be proved. It is plausible that his renal lesion and subsequent complications might have resulted from such a primary metabolic magnesium abnormality. Rapado et al. (1975) and Rapado and Castrillo (1976/1980a,b) have reported patients with chondrocalcinosis and renal calcinosis who had magnesium malabsorption.
It is of interest that calcium, vitamin D, and often PTH were used to control the neuromuscular irritability, associated with the first diagnosed hypocalcemia, in almost all of the cases cited. Their serum calcium rose, sometimes to hypercalcemic levels, as did their serum alkaline phosphatase. Their serum phosphorus levels dropped without improving the serum magnesium levels or the clinical signs, until their magnesium deficiency was diagnosed and corrected. Other children, who had histories of clinical signs suggestive of hypomagnesemic hypocalcemia, developed hypercalcemia when magnesium therapy was added to their high-dosage calcium and vitamin D therapy on which they were being maintained to control their hypocalcemia. These observations suggest that their release of PTH (Review: Anast, 1977), its conversion to an active form (Passer, 1976), or response to vitamin D may have been abnormal in the presence of hypomagnesemia. That magnesium therapy increases the calcemic response to vitamin D in hypoparathyroid patients has been recognized for many years.
Two of the infants described in this section were hypocalcemic, not responding to high-dosage vitamin D (Stromme et al., 1969; Nordio et al., 1971). Whether these infants had vitamin-D-resistant rickets that failed to respond to very high doses of vitamin D until their magnesium deficiency was repaired seems possible. Magnesium-dependent vitamin-D-resistant rickets has been described in a hypoparathyroid girl (Rösler and Rabinowitz, 1973) and in a rachitic boy (Reddy and Sivakumar, 1974), both of whose serum calcium levels rose and vitamin D requirements dropped substantially on correction of hypomagnesemia. It should be noted that the use of high doses of calcemic agents to raise the blood calcium of hypophosphatemic vitamin-D-resistant rickets (Moncrieff and Chance, 1969) or of other hypomagnesemic hypocalcemias can be nephrotoxic. Thus, evaluation of children with abnormal requirements or response to vitamin D for their magnesium status is indicated. Since vitamin D is necessary for the absorption of magnesium, children with abnormal vitamin D metabolism might have concomitant magnesium deficiency. Among those with hyperreactivity to vitamin D (Review: Seelig, 1969b), the excess magnesium loss (caused by hypervitaminosis D) can cause renal and cardiovascular damage directly, to which the vitamin D excess is contributory.
All but one of the affected infants were boys. In several of the families (Stromme et al., 1969; Salet et al., 1970; Bardier et al., 1970; Vainsel et al., 1970), more than one child was affected. In another (M. Friedman et al., 1967) the parents were closely related. The genetics of abnormalities in magnesium intestinal absorption needs evaluation. Determination of the incidence of marginal magnesium deficiency in parents, siblings, and other close relatives of children with primary magnesium malabsorption should be ascertained, as should possible relationships with abnormalities in vitamin D metabolism.
Calculation of the amount of magnesium infants must retain daily (0.85 mEq) for normal growth and development and for their metabolic processes suggests that they have only a narrow margin of safety, assuming normal intestinal absorption (Harris and Wilkinson, 1971). Thus, infants are particularly subject to magnesium depletion when they have acute or protracted diarrhea. Breton et al.(1961), considering that the stool of infants with severe acute diarrhea contains an amount of magnesium almost equal to that ingested (Holt et al., 1915), noted that the distribution of serum magnesium levels of such infants and of those with chronic diarrhea of mucoviscidosis did not differ substantially from that of normal infants. During recovery, however, the serum magnesium levels gradually fell, an observation that they considered suggestive of hemoconcentration during intestinal loss of fluid and of possibly transitory renal insufficiency, which masked the actual (tissue) deficit. In more severe gastroenteritis with refractory vomiting and diarrhea, severe hypomagnesemia has been reported (Back et al., 1962). Among 5 infants (8 months to 2 years of age), with symptomatic hypomagnesemia associated with gastroenteritis, there were 3 with protein calorie malnutrition (infra vide) and 2 (1 and 2 years old, respectively) whose magnesium deficit seemed to be the result of the disturbance of the alimentary tract. Both infants improved on magnesium therapy, after i.v. fluids had proven ineffective and i.v. calcium had superimposed convulsions on the tetanic state. The authors noted the importance of magnesium in general cellular metabolism, and the observations of R. Fletcher et al. (1960) that repair of the deficit may improve impaired intestinal function. All 20 infants, who were severely dehydrated as a result of severe gastroenteritis and who had received intravenous therapy, developed one or more neurologic manifestations of magnesium deficiency (Back et al., 1971/1973). Eleven had been well nourished before the acute episode; all recovered. Nine had been malnourished; 7 died. Of the 12 infants who developed neurologic signs only after intravenous therapy had been started, nine were being given additional potassium at the time. They reported that in their series of 20 children admitted with severe gastroenteritis all 20 developed neurologic signs and symptoms of magnesium deficiency, 75% while they were receiving potassium therapy. They stressed that it is important to correct both deficiencies, and found that when magnesium was given parenterally as magnesium sulfate (2 ml 25% solution) 15 of the 18 infants so treated responded with correction of their symptoms within 10 minutes; symptoms did not recur. Two had only partial response. One was also given calcium gluconate to control carpopedal spasm, but when the calcium was given again five hours later to control convulsions, it was ineffective; magnesium therapy controlled the convulsions.
Prolonged gastroenteritis in a three-month-old infant, starting a month after a colostomy had been performed for intestinal obstruction, was followed by feeding full-strength vitamin-D-fortified cows' milk formula when the acute problem was corrected (Savage and McAdam, 1967). Clonic convulsions developed, which were found to be caused by hypomagnesemia (0.54 mEq/liter). The authors attributed the magnesium deficiency to a combination of factors: prolonged gastroenteritis (causing losses of magnesium and calcium), large feedings of full-strength cows' milk formula (replacing predominantly the calcium), and rapid growth during convalescence (increasing magnesium requirements). They cautioned that it might be unwise to give cows' milk to an infant recovering from severe diarrhea without first checking the magnesium and supplementing if indicated.
As in infantile severe diarrhea, which is accompanied by dehydration (Breton et al., 1961; Paupe, 1971), the magnesium status of patients with cholera is difficult to evaluate and precarious. Kobayashi (1971) commented that muscle cramps and convulsions were often encountered during the rehydration phase, particularly in children, and in those given physiologic saline and sodium bicarbonate rather than lactated Ringer's solution. They reported that during the acute phase of the disease, hypermagnesemia (2.68-3.75 mEq/liter) was not uncommon, although patients under six years of age had mean levels of 2.68 mEq/liter ± 0.56.
Paupe (1971) reviewed the contribution of acute and chronic diarrhea in infancy and childhood to hypomagnesemia. He pointed out that such deficits might be missed, on measuring serum magnesium levels, because of the dehydration associated with loss of gastrointestinal fluids. On the other hand, failure to compensate for magnesium losses is likely explain the transitory and marginal hypomagnesemias reported during convalescence from acute diarrhea (Breton et al., 1961; Bernal et al,, 1967). To avoid losses sufficient to be reflected by hypomagnesemia, Harris and Wilkinson (1971) administered magnesium salts to such infants empirically for many years with favorable results. They employed the procedure to determine magnesium depletion by ascertaining the percentage urinary retention of a parenteral load of magnesium, and showed that 20 to 29 infants suspected of magnesium depletion retained over 40% of the load. In 16 infants with established magnesium depletion, the most frequent cause was frequent watery stools. One of the patients with serum magnesium levels above the normal range (1.4-1.9 mEq/liter) had a low muscle magnesium level (1.17 mEq/liter; normal = 1.63-2.35) and retained 50% of the test dose. The serum magnesium levels were normal in three who were shown to be magnesium deficient by their retention of more than 70% of the test dose. Not only were the signs of irritability or convulsions improved by the magnesium, but the diarrhea itself showed improvement that seemed related to the magnesium administration. This observation is of particular interest in view of the report by Woodard et al.(1972) that an infant with selective malabsorption of magnesium had secondary diarrhea that remitted on repletion of magnesium.
The malnutrition seen in infants and young children, kept breast-fed too long to avoid the risk of gastroenteritis encountered on adding food prepared and kept under unhygienic conditions in undeveloped countries, has been termed kwashiorkor or protein calorie malnutrition (PCM) (Frenk, 1961). Affected children are usually one to four years of age and are generally hospitalized in grave condition after periods of protracted diarrhea, often vomiting, and usually with muscle wasting, dehydration, and trophic disturbances of the skin. There are many variations in therapeutic approaches to the emergency situation, which entail immediate correction of the dehydration by intravenous infusion, followed by skim milk (often protein-fortified), potassium, iron, vitamins, and cottonseed oil (Dean and Skinner, 1957). "Recovery syndromes," with edema, neuroirritability, and (in some geographic areas) cardiovascular abnormalities have been described (Frenk, 1961; Caddell, 1965, 1969a; Wharton et al. 1968), and have been attributed to nutritional imbalances that become manifest, or even provoked by the therapeutic regimen.
Such infants are particularly susceptible to development of hypomagnesemia and tissue depletion of magnesium, particularly when the therapeutic regimen is not only low in magnesium but high in calcium, phosphate, and protein, which lead to new tissue formation and increased magnesium requirements. The first hint that babies (in Uganda) with PCM might have an abnormality in their magnesium metabolism was provided by Schwartz (1956), when she correlated low serum alkaline phosphatase levels of infants with their failure to grow, and showed that their plasma enzyme activity could be increased in vitro by addition of magnesium. Standard treatment (in India) lowered alkaline phosphatase activity twofold from levels on admission (Mukherjee and Starker, 1958), an observation that further indicates that such a diet might have intensified the magnesium deficiency (Caddell, 1965). Low muscle levels of magnesium in Mexican children with PCM were correlated with blocks in aerobic glycolytic metabolism at Mg-dependent enzymatic steps, e.g., those involving pyruvate and alpha-ketoglutarate metabolism (Metcoff et al., 1960, 1963).
The first demonstration of improvement in clinical response of babies with PCM when magnesium was added to their regimen to correct their hypomagnesemia and low skeletal muscle magnesium levels was in Jamaica (Montgomery, 1960). His magnesium balance studies in such children the following year showed retention of about half the magnesium supplements, even while diarrhea continued. Such additions to the standard regimen resulted in rises in muscle magnesium and potassium, fall in muscle sodium, and improvement in edema (Montgomery, 1961b). His group then showed that the neurologic manifestations of hypomagnesemia and hypocalcemia of severe PCM responded to treatment with magnesium, but not to calcium alone (Back et al. 1962). They later found that, although in some instances the muscle potassium deficit might be even greater than that of magnesium in some PCM children (Alleyne et al., 1970), treatment that corrected the potassium deficit without simultaneously meeting the magnesium needs might have adverse effects (Back et al., 1971/1973). The neurologic signs of 15 of the 20 infants and children with severe gastroenteritis, with and without PCM, developed while they were receiving potassium therapy. They observed that since potassium loads to magnesium-deficient animals precipitate neurologic signs, it would be prudent to correct both deficits clinically. It was noteworthy that seven of the nine infants in their series of 20 who had been severely malnourished did not survive, that their CSF magnesium levels were subnormal, and that they had cerebral edema on autopsy. Magnesium repletion (2 ml 25% MgSO4 given parenterally, controlled the neuromuscular irritability in 15 of 18 of the infants who had both deficits corrected.
The importance of these findings is indicated by the observations of Wharton et al. (1968) that despite lack of agreement as to the best mode of treatment of PCM, all therapeutic regimens include potassium. Although most studies have confirmed the observations of Montgomery (1960, 1961a) and Metcoff et al. (1960) that magnesium and potassium losses in muscle of children with PCM usually parallel one another, there has been controversy as to whether magnesium supplements improve the prognosis of children with PCM undergoing treatment. Wharton et al. (1968) point out that some of the differences in clinical manifestations of the disease, and in response to therapy, may reflect geographic differences, both in dietary conditions and therapeutic preferences. They point out that in Uganda and Nigeria, cardiovascular complications during nutritional repletion are a considerable risk, while in Jamaica pulmonary edema and hepatic failure are more common; in both those areas and in Central America and India, peripheral edema and neurologic abnormalities are common. It was in Central Eastern Africa that correction of the demonstrated magnesium deficit, precipitated by the standard therapeutic regimen, was shown to reverse the resultant electrocardiographic abnormalities, as well as improve the edema and neurologic status and both morbidity and mortality (Caddell 1965, 1967, 1969a,b; Caddell and Goddard, 1967). All six of the magnesium-supplemented children in the initial study in Uganda (Caddell, 1965) survived; 12 of 21 on the standard regimen died. Extension of her studies in Nigeria (Caddell, 1967, 1969b) showed comparable neurologic and cardiovascular changes among the children on the standard regimen: high-protein milk plus vitamins and minerals (low in magnesium). All 13 Nigerian children with PCM, who had skeletal muscle tissue analyses, showed low levels of magnesium; hypomagnesemia was present in 18 of the 27 children tested (Caddell and Goddard, 1967). The double-blind paired sequential study of 52 severely malnourished Nigerian children, none of whom had shown much improvement on rehydration therapy, was performed to determine the extent to which addition of magnesium to the customary regimen would improve the therapeutic response (Caddell, 1967). The children who received parenteral magnesium could be distinguished from those given equal volumes of isotonic saline by the rise in subnormal temperatures and blood pressures within 24 hours, and general improvement in five days. Fifteen of the 26 magnesium-treated children showed remarkable recoveries. Three died early, and nine developed serious infections, from which only one recovered. In contrast, half of the 16 control children died, three early, two of unknown cause after temporary improvement. Eight died among the 21 who were found to be in the control group when the code had to be broken because of worsening clinical condition; the remaining 13 made remarkable recoveries on substitution of magnesium sulfate for the saline injections. Because magnesium is a hypotensive agent, it had been withheld from the first three children who became hypotensive; they died despite administration of vasopressors and blood transfusions. Later, when it was realized that the magnesium deficiency might be contributory to the hypotension, magnesium therapy was cautiously instituted, sometimes with dramatic improvement. A later report (Caddell, 1969b) considered the susceptibility of PCM children on standard therapy to congestive heart failure, when given blood transfusion, and their subsequent high incidence of digitalis toxicity. During a 2- to 12-month follow-up period of 32 children who survived their severe PCM, and who had received parenteral magnesium, Caddell (1969a) contrasted the sustained rapid improvement in her series of patients, with the persistent abnormalities and stunting for prolonged periods of time, and high mortality rates among the PCM children who had not received magnesium supplements reported by others.
Evidence of magnesium depletion in children with PCM, comparable to that seen in Uganda and Nigeria, has been reported from Senegal (Ingenbleek and Giono, 1971/1973). Hypomagnesemia (mean = 1.1 mEq/liter) was noted on admission in the 11 babies whose magnesium and nitrogen balances were studied for 23-30 days, while they were on oral magnesium supplements (240 mg Mg/48 hours). Cumulative magnesium retention reached about 2 g. This group of investigators had found, earlier, that the signs of neuromuscular irritability had been intensified after the first week or two of protein repletion in a small percentage of severely ill babies, during which time their initially strongly positive magnesium balance dropped sharply. When the diet was gradually improved and supplemented with magnesium, the magnesium balances steadily became more positive and the "recovery tetany syndrome" did not develop. Unlike the repletion of potassium, which had been accomplished by 10 days of oral potassium chloride, independent of the rate of nitrogen retention (Ingenbleek et al., 1968), the magnesium repletion seemed linked with that of nitrogen retention.
The dietary staple (maize) among the Bantu in South Africa being richer in magnesium than in the main dietary constituent in Central Africa, cassava (Rosen, 1971), it is not surprising that the magnesium deficiency of children with PCM in that area has been less severe, and that their responses to magnesium supplementation less striking. Linder et al., (1963) found that Bantu infants with PCM fed skim milk alone, supplemented by infusions to combat dehydration when necessary, produced positive magnesium balances. However, five of the children, who were also given 130 mg of magnesium daily retained twice as much magnesium as did the patients without the supplement. Those given magnesium also retained more calcium than did those on milk alone. The mean serum magnesium levels of the babies given magnesium supplements reached almost normal levels within the first nine days of treatment; the mean levels of those receiving no supplement remained at 1.2 mEq during the same period. From days 10-22, the mean serum magnesium levels of the supplemented babies reached 1.6 mEq/liter; that of those without supplements rose to 1.4 mEq/liter. Thereafter, there was little difference in serum levels in the two groups. Pretorius et al. (1963), also in South Africa, found that babies with PCM did not have serum and erythrocyte levels of magnesium as low as did Jamaican babies with PCM (Montgomery, 1960, 1961a). Nonetheless, they retained up to 60% of parenterally administered magnesium, very small amounts of which appeared in the urine. They had malabsorption of magnesium that persisted, even after diarrhea had abated. Rosen et al.(1970) did not confirm Caddell's (1967) findings of improved therapeutic response in their South African study of 100 consecutive children with PCM, 50 assigned to the standard regimen and 50 to the same basic regimen plus magnesium supplementation. The mortality rates in both groups were 21% (most early after admission) and the rates of recovery were the same. The serum magnesium levels were slightly lower than normal in the children with PCM, but the differences in incidence of low and high in the groups on standard and magnesium-supplemented regimens did not differ substantially. This group (Rosen et al., 1970) did not have to break the code before completion of the study because of worsening clinical condition, as did Caddell (1967). Unlike the children in Nigeria and Uganda, electrocardiographic changes that improved when magnesium was added were not part of the recovery syndrome on the standard regimen in South Africa.
Studies from India have confirmed the magnesium depletion of young children with PCM (Agarwalet al. 1967; Bajpai et al., 1970; Chhaparwal et al., 1971a; S. Mehta et al., 1972). Although low blood magnesium levels have been reported frequently, serum and erythrocyte levels did not always reflect the status of magnesium in the body. Bajpai et al. (1970) compared the levels of magnesium in plasma, erythrocytes, and skeletal muscle in children with PCM and in a group of children, some of whom were convulsing from "minor ailments" (Table 4-5). Since magnesium deficiency can be implicated in pediatric convulsive states, it is uncertain that the range for the controls reflects optimal magnesium levels. The magnesium blood levels of the PCM children who had diarrhea were lower than in those without diarrhea, an expected finding in view of the magnesium deficiency caused by inflammatory or metabolic intestinal disease. Almost a third of the children with PCM had plasma magnesium levels below 1.40 mEq/liter; half had erythrocyte magnesium levels below 3.50 mEq/liter packed cells. The muscle magnesium levels were 30% lower in the PCM children than in the three controls whose muscles were biopsied. Comparison of magnesium levels of eight children given parenteral magnesium (1-1.5 ml, 50% MgSO4 for three days and then 1 ml on alternate days) for 18-20 days (randomly allocated) and those not so supplemented showed that the magnesium supplemented children exhibited significantly increased erythrocyte magnesium levels, as compared with those getting standard therapy. Only slight increases in plasma and muscle magnesium levels were noted in those getting magnesium; three of four patients (not on magnesium) who were again biopsied showed decreased muscle magnesium levels. The minimal increases in muscle magnesium, even in those being supplemented, suggested to the investigators that the demand outstripped the supply.
{kind=link}
Caddell's investigations of PCM children in Thailand provide evidence of the difficulty in selecting laboratory tests that reliably indicate magnesium depletion in such children. In the first of these studies (Caddell and Olson, 1973), 44% of the 30 children had serum magnesium levels just below the lower limit of normal at the time of admission, but 93% had significantly low urinary Mg outputs; muscle magnesium levels were almost half the published normal value. After 16 hours of parenteral fluids that had no magnesium, 56% of the plasma Mg levels had decreased. The lowest plasma magnesium values developed between days 5 and 14, when plasma albumin and other electrolytes were attaining normal levels. Anorexia persisted longer in the children with plasma levels of magnesium below 1.2 mEq/liter than in those with higher levels. Pitting edema, T-wave abnormalities, and neurologic signs and symptoms correlated with levels below 1.0 mEq/liter during the early treatment period. Continued diarrhea, prolonged intravenous therapy, and anorexia were contributing factors to the drop of plasma magnesium levels to 1.0 mEq/liter in 17 children who were being treated with magnesium. Normal plasma magnesium levels were attained in all but one child by three weeks, but almost one-third still had low urine magnesium values. There was little difference between 24-hour urinary outputs of magnesium in those receiving and those not receiving magnesium supplements; both groups had hypomagnesiuria. Muscle magnesium levels increased slowly and were still low at 11 weeks. In the second study of Thai children with PCM (Caddell et al., 1973), the parenteral magnesium load test was utilized to provide a better clue to the magnesium status of these malnourished children, before and in the course of nutritional repair. Low preload urinary magnesium excretion was not found a helpful guide in this series of children, who had relatively mild hypomagnesemia. Seven of 25 children, who excreted less than 1 mEq/liter/24 hours, retained a mean of only 23% of the magnesium load. There was no significant correlation between magnesium retention and edema; children with antecedent diarrhea retained much of the magnesium load.
Aguilar (1971/1973) and Cheek et al. (1970) found that Peruvian PCM children retained both magnesium and potassium, in proportion to that of nitrogen, but that the minerals were more quickly retained than was the nitrogen. Low muscle magnesium levels were found in the PCM children before and four to nine months after treatment (Cheek et al., 1970). In a detailed metabolic study from Guatemala (Nichols et al., 1978), it was found that increasing the daily oral magnesium supplement to 0.42 mEq/kg/day, from the amount (0.12 mEq/kg/day) that had been found insufficient for adequate retention (Nichols et al., 1974), resulted in five to six times greater magnesium retention and markedly increased muscle magnesium levels. Provision of 2.7 mEq/kg/day (from oral and parenteral supplements of magnesium) was not essential for clinical recovery from the edematous form of PCM, but their response was more rapid than was that of those on the lower supplements. Their muscle potassium levels returned to normal earlier, and on a constant intake their potassium retention was increased threefold during the magnesium supplementation. Considering the insensible losses of magnesium (i.e., from skin), the amount required for restoration of deficit, and that needed for formation of tissue, Nichols et al. (1978) estimate that the oral magnesium requirement during initial stages of treatment of PCM may be as high as 2.7 mEq/kg/day (32 mg/kg/day). When diarrhea interferes with absorption, combination of parenterally administered and oral magnesium is necessary.
Aguilar (1971-1973) made an interesting observation on the failure of the kidneys of the infants with PCM to retain magnesium when their Mg supplements were discontinued. Whether this implies tubular damage like that found in magnesium deficient rats (J. Oliver et al., 1966) and in an infant with primary malabsorption of magnesium (Vainsel et al., 1970) remains to be determined. Renal damage in the area where magnesium is reabsorbed may intensify magnesium deficiency or make repletion difficult (Seelig et al., 1979). It has been shown, however, that children with PCM have subnormal renal function (Nichols et al., 1974).
There are few more tragic events than the sudden death of an infant who seemed healthy, was growing well, and had few signs of anything wrong more serious than a slight respiratory infection, irritability, or feeding difficulties a day or two before being found dead in his crib. Research into the literature has disclosed such events throughout history; epidemiologic studies and reviews show that they are most common in the winter to spring months and most often occur in infants of young and multiparous mothers (Valdes-Dapena, 1967; Geertinger, 1967; Froggatt et al., 1968; Marshall, 1972). The incidence is about 1 in 400-500 live births; over 20,000 are estimated to occur annually in the United States (Froggatt et al., 1968; Valdes-Dapena, 1973). The etiology of such deaths remains undefined. Asphyxiation and parental neglect used to be blamed. The major theories now include (1) viral infection and immunologic abnormalities or histamine shock (Froggatt et al., 1968; P. Gardner, 1972; Caddell, 1972; Ogra et al., 1975; Caddell, 1975/1977); (2) disorders of the autonomic system (Salk et al., 1974; Naeye, 1976; Naeye et al., 1976a) that can lead to periods of apnea, such as are frequently implicated in SIDS (Steinschneider, 1972; Naeye, 1973; Guilleminault et al., 1975); and (3) abnormalities in cardiac conduction tissue and electrical instability of the heart (T. James, 1968; J. Ferris, 1972, 1973). Swift and Emery (1972) question the histamine theory, since they found no degranulation of pulmonary mast cells in SIDS victims. Also controversial is the theory that such infants have abnormal conduction tissue (Valdes-Dapena et al., 1973; Lie et al., 1976; T. James, 1976). A fortuitous study of cardiac lability in a group of healthy infants, one of whom later died suddenly, showed that the prestimulus variability in heart rate of the SIDS infant was significantly deviant from the other 23 infants subjected to auditory stimuli; his peak accelerated rate was higher (Salk et al., 1974). Increased muscle mass of the pulmonary arteries have been detected in SIDS victims, and considered a possible consequence of chronic alveolar hypoxia that might reflect the periods of sleep apnea (Naeye, 1973; Naeye et al., 1976a,b).
The possibility that magnesium deficiency of growth might be a major factor in the etiology of SIDS has been postulated by Caddell (1972). She points out that premature and low-birth-weight infants with poor magnesium stores and low birth weights are most vulnerable to sudden unexpected death. She reviewed the evidence that this condition used to occur in breast-fed infants of destitute multiparous mothers, but that it is now chiefly a problem of infants (often overweight) fed artificial formulas and cereal foods that provide high contents of calcium, phosphorus, and protein. The development of hypomagnesemia in formula-fed infants, often in association with hypocalcemia despite the higher content of calcium in cows' than in human milk has been discussed earlier. Formula-fed infants commonly grow faster than do breast-fed infants, and have lower plasma magnesium, as well as calcium levels, and higher phosphorus levels. Thus, they may well fit into Caddell's (1972) "magnesium deprivation syndrome of growth," particularly when they are born to mothers whose magnesium status may be suboptimal or poor, and thus might have insufficient magnesium stores at birth. Maternal hypomagnesemia has been demonstrated in precisely those women whose infants are at greatest risk of SIDS: women with preeclampsia or eclampsia, who are themselves immature and whose diets do not meet their own growth requirements of magnesium, or who are of high parity, particularly when the pregnancies have been at frequent intervals. Infants born to such mothers, especially if the birth is multiple, probably have low magnesium stores. Support for this premise derives from the observation that the young of magnesium-deficient pregnant animals are more magnesium deficient than are the mothers (Cohlan et al., 1970; Dancis et al., 1971; Wang et al., 1971) holds true for human infants. Caddell (1972) has pointed out that premature infants, whose magnesium stores are proportionally less than are those of a full-term infant (Widdowson, 1965), and who have high growth rates, might reach critically low levels of magnesium that might trigger the SIDS. She compares their premonitory and terminal signs to those of acute magnesium deficiency in immature animals and to those in infants recovering from severe gastroenteritis of protein calorie malnutrition, among whom sudden death has been reported during the recovery period, at which time new tissue formation increases magnesium requirements.
One may question whether the "sniffling" or signs of a minor respiratory ailment, which is commonly reported as a premonitory sign of SIDS, is the human counterpart of the reddened, inflamed snout and ears of magnesium-deficient animals. Such reactions might reflect histamine release, and magnesium deficiency has indeed been shown to increase degranulation of mast cells and to increase histamine blood and urinary levels (Hungerford and Karson, 1960; Bois, 1963; Bois et al 1963; Bois and Jasmin, 1971/1973). The similarity of some of the SIDS necropsy findings to those of anaphylactic shock, with hemorrhagic and edematous pulmonary changes, supports Caddell's hypothesis that the sudden death might be mediated by release of histamine
Although neuroirritability is common a day or two before the sudden death (Caddell, 1972), the typical picture of acute experimental and clinical magnesium deficiency, seizures and electrocardiographic changes, is usually not characteristic of the SIDS. Tonic-clonic seizures have been reported in SIDS, but a retrospective survey of the temperament of victims of SIDS provides evidence of less intense reactions to environmental stimuli than had been exhibited by normal siblings (Naeye et al., 1976a). They were less active, more often breathless and fatigued, and had more shrill cries. A prospective study found additional evidence of central nervous system dysfunction, including neonatal abnormalities in respiration, labile temperature regulation, and weak suck reflexes. Despite the commonly held assumption that the SIDS strikes infants who were completely well before the catastrophe, Naeye et al. (1976a), obtained evidence that only a third of the SIDS victims were completely free of illness or unexplained crying.
Just as magnesium deficiency can be implicated in histamine release (as a contributory factor to the SIDS), perhaps a less acute magnesium deficiency might also be involved in cardiac changes, described in the conduction tissue of infants with the SIDS, and that can cause sudden death as a result of acute arrhythmias. Magnesium deficiency and agents that increase myocardial magnesium loss have been utilized in many experimental models of myocardial necrosis (Reviews: Lehr, 1969; Seelig, 1972; Seelig and Heggtveit, 1974). As in those models, infantile coronary arteriosclerosis generally involves the small intramyocardial arteries, with perivascular foci of infiltration, necrosis, and fibrosis. If the areas of necrosis involve the conduction tissue, even small foci can induce arrhythmias and sudden death (T. James, 1967). The high lability of magnesium in the interventricular septum and left ventricle (p.187) suggest that these areas are at particular risk in infants with suboptimal magnesium. It is not yet clear whether small coronary lesions contribute to damage to the conduction system of the heart in the SIDS (T. James, 1968; Ferris, 1972, 1973; Valdes-Dapena et al., 1973; T. James, 1976). W. Anderson et al. (1970) found focal intimal and medial hyperplasia of the A-V node artery with luminal narrowing in 35% of the SIDS cases and in 10% of 22 control infants of the same age (between one and two months). However, resorptive and degenerative changes involving portions of the A-V node and bundle of His was present in all SIDS and control cases. They speculate that dysfunction associated with these processes might be contributory to the SIDS. Ferris (1973) has commented that the changes in the conductive tissue of the heart of infants with the SIDS is akin to the form of ischemic fibrosis that is seen with adult coronary arterial disease. Valdes-Dapena et al. (1973) observed petechiae in the conduction system of 26% of SIDS infants and 20% of control infants in their group of 47 who had died in the first year of life, an insignificant difference. They noted that 50% of the 31 STDS infants had minute myocardial hemorrhages and that 37% of the 16 controls had similar hemorrhages in the myocardium near the conduction system. However, they disagreed that there were connective tissue changes near the conducting system that might explain the sudden deaths. It should be noted that the myocardial hemorrhages described in both groups seem to indicate some abnormal process; that they occurred in both groups might reflect a common underlying abnormality. Among the control infants were 6 with pulmonary disease (infection or hyaline membrane), 1 with methemoglobinemia, 1 who was premature, and 2 with diseases causing severe diarrhea; all are conditions that might well have caused loss of myocardial magnesium.
4.7.1.3. SIDS and Hypoparathyroidism
Another condition that has been directly associated with the SIDS is infantile hypoparathyroidism, a condition associated with maternal hyperparathyroidism and with neonatal hypomagnesemia, hypocalcemia, and hyperphosphatemia. The study of 82 autopsied cases of SID (Geertinger, 1967) showed that in a third of the infants, no parathyroid gland could be found. In the others there were abnormalities in parathyroid localization and morphology, often with fusion with thymic tissue. The author speculated that maternal hyperparathyroidism might result in congenital anomalies of the parathyroids. Thus, the experimental model that might be most relevant to the cardiac damage of infants in the first few months of life, and consequently to the SIDS and to other sudden deaths and cardiac lesions during infancy, is the parathyroidectomized, phosphate-loaded rat that develops lesions of the small coronary arteries and of the perivascular myocardium (Lehr, 1959, 1965). Neonatal infants are commonly hypoparathyroid, and those fed cows' milk formulas are also hyperphosphatemic. Those born with poor magnesium stores are particularly vulnerable to lesions of the small coronary arteries, such as have been produced in "pure" magnesium-deficient animals, and intensified by phosphate loads (Review: Seelig and Haddy, 1976/80).
Hyperparathyroidism in the mother, which predisposes to infantile hypoparathyroidism, might be the result of maternal hypomagnesemia. Resultant mobilization of maternal calcium, and its transfer to the fetus, militates against fetal hyperparathyroidism, and has in fact been implicated as the cause of infantile hypoparathyroidism, which is often associated with hypomagnesemic hypocalcemia. That infantile hypomagnesemia can be associated with congenital absence of the parathyroids and thymic abnormalities [such as Geertinger (1967) showed in the SIDS], has been reported by Taints et al.(1966) in an infant with neonatal tetany associated with persistently low serum magnesium levels. Niklasson (1970) reported two sisters with similar manifestations and hypomagnesemic hypocalcemia in a family with a high incidence of hypoparathyroidism. Eight members of the family had died during infancy, one at four weeks of "sudden unexplained death" and four with convulsions at under six months.
The role of hypomagnesemia in refractory hypocalcemia of infancy suggests that, in addition to the association of hypocalcemia with recurrent apnea of premature infants (Gershanik et al., 1972), the magnesium status should also be ascertained. The investigators (Gershanik et al., 1972) found no difference in the overall mean magnesium levels between the infants who did or did not suffer attacks of apnea. In view of the egress of magnesium from cells, however, in response to hypoxia the normal serum magnesium levels in infants with recurrent apnea cannot be accepted as proof that magnesium deficiency was not present. Measurement of retention of a parenteral magnesium load would provide a more reliable index of the infants' magnesium status (Harris and Wilkinson, 1971; Caddell, 1975).
Far from all infants who die suddenly are autopsied; many are classified as SIDS on the basis of the clinical history, no clear medical explanation for the death having been noted. However, only a third of the SIDS infants had had no premonitory signs (Naeye et al., 1976a). Intensive interviews with their parents disclosed that most had tended to be more subject to breathlessness and exhaustion during feeding than were their siblings, and to have less reactivity to environmental stimuli. These manifestations are not unlike those reported for infants found at autopsy to have cardiovascular lesions, such as coronary artery disease (with or without myocardial infarcts), endocardial fibroelastosis, or both, and who-although they often died suddenly-are thus not included in the SIDS category. (Sudden death was reported in about one-fourth of the infants reported in Appendix TablesA-5A and A-6A.) Their prodromal symptoms, however, resemble those described in SIDS. Sudden onset of respiratory distress in previously well-nourished, thriving infants was the presenting finding in many of the infants found to have coronary disease, endocardial fibroelastosis, or focal myocardial lesions at autopsy. Cyanosis and intermittent episodes of pallor and cold sweats were common. Most died within a few hours to a few days after the onset of the sudden illness. Many of the infants also presented with feeding difficulties and vomiting, often of sudden onset. ECU tracings typical of ischemic heart disease were sometimes obtained.
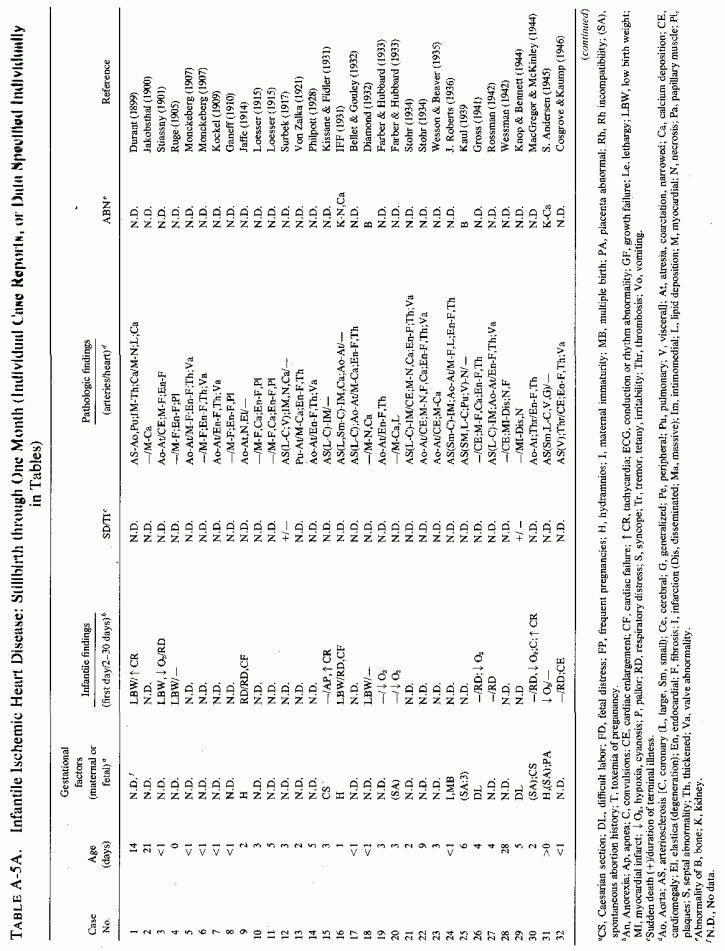{kind=link}
{kind=link}
4.7.1.4. Epidemiologic Factors in SIDS
Since magnesium deficiency has been implicated in sudden death from ischemic heart disease in adults, the incidence of which is much higher in soft-water areas with low magnesium content than in hard-water areas (T. Anderson et al. 1975, 1979), and since a highly significant negative correlation has been found between infant mortality and water hardness (M. Crawford et al., 1968, 1972), it may be that magnesium deficiency in soft-water areas is contributory to the SIDS and to diagnosed infantile cardiovascular disease. This group noted that the correlation was much higher in the 1968 and 1972 studies than it had been in a 1951 analysis, and proposed that water minerals might play an important role in infant mortality that became manifest as "social" factors became less important. In 1972, M. Crawford et al. selected older mothers and those of high parity as being at high risk for both stillbirths and infant deaths, and found that the highest incidence of stillbirths and postneonatal infant deaths occurred in women of parity 3+ and in areas with the softest water. They speculated that it was the low calcium level in the soft water that was the risk factor. They did not include magnesium determinations in the 1972 study, but in the 1968 study gave data showing that the magnesium level in soft-water communities was about a quarter that of the hard-water communities.
Studies from Finland provide further data that suggest that it might be the amount of magnesium consumed that influences susceptibility to sudden death from ischemic heart disease, not only in adults but in infants. Karppanen and Neuvonen (1973) pointed out the clear-cut regional distribution of ischemic heart disease in Finland, being twice as high in eastern as in southwestern Finland. It is thus of interest that the magnesium content in the east Finland soil is one-third that of southwest Finland. A study of the thickness of the inner layers of the coronary arteries of infants showed that infants from families from the eastern parts of Finland had significantly thicker coronary arteries than did those from the southwest, a finding correlated with a higher rate of adult ischemic heart disease in the eastern part of the country than in the southwest (Pesonen et al., 1975). There has also been a report from Finland of infant death in the first three children born to consanguinous parents (Meurman et al., 1965). The first died one hour after birth, the victim of birth asphyxia. The second thrived until six weeks, at which time she suddenly refused her feedings, had screaming attacks, and died before she could be hospitalized. Autopsies were not performed. The third infant developed identical symptoms to that of the second, at six weeks of age, and died suddenly at night. She had the typical coronary lesions of infantile arteriosclerosis. This family lived in Kuopio, in the northeastern part of Finland. Possibly contributory might be frequency of pregnancies in the presence of suboptimal magnesium intake. However, at the time of the publication, the fourth and fifth children were well at two years and at four months, respectively. Follow-ups of these infants are not available.
There is an overlap in the months during which the greatest number of infants die with the SIDS and in which most cases of infantile tetany have been reported. A survey of the world literature showed that there were twice as many cases of SIDS during the colder months of the year (Valdes-Dapena, 1967), a finding confirmed by an epidemiologic survey in Ireland indicating that the peak incidence occurred between February and March (Marshall, 1972). It is provocative, thus, that the serum calcium levels were low during gestation in the winter months (Mull and Bill, 1934) despite their hyperparathyroidism (Bodansky and Duff, 1939), and that neonatal tetany, a condition that is correlated with transient hypoparathyroid ism and hypomagnesemic hypocalcemia, has also been shown to be most frequent in the cold months (Saville and Kretchmer, 1960). The possibility that hypoparathyroidism might be implicated in the SIDS (Geertinger, 1967) has been considered, as has the possible role of parathyroid deficiency in damage to small coronary arteries and in perivascular myocardial necrosis. Ludwig (1962), who reviewed the status of infants born of hyperparathyroid mothers, found of the 40 infants reported (presumably hypoparathyroid, at least at birth) there were 9 who were stillborn or aborted, 5 who died shortly after birth, and 5 who developed neonatal or later tetany. Five of the infants were premature.
Magnesium determinations are almost never reported in mothers or siblings of infants who died of the SJDS or of proved cardiac failure or arrhythmias, or in infants with congenital cardiovascular disease. Convulsions, the condition that most often leads to such tests, are rarely part of the prodromata of infants with these disorders. Although low-birth-weight infants, multiple births, those born to diabetic mothers or to multiparous mothers have been evaluated for serum magnesium levels, there is a paucity of follow-up data as to the incidence of the SIDS or cardiovascular disease in such infants. Because hypoxia causes egress of magnesium from the tissues, serum magnesium levels might provide unreliable assurance of normal magnesium body levels in infants with cardiac failure. Determination of the magnesium status by ascertaining the percentage retention of a loading dose is of value if the renal function is normal. Improved techniques are necessary for evaluation of cellular levels of functional magnesium. Caddell and her colleagues are addressing themselves to a systematic survey of the SIDS problem, attempting to determine whether maternal magnesium deficiency, as determined by retention of load-test, is participatory (Caddell, 1975, 1977; Caddell et al. 1975). Similar surveys of mothers and siblings of infants who died of coronary arteriosclerosis and other cardiac lesions, as well as of babies with congenital cardiovascular disease, is also indicated.
No comments:
Post a Comment