

MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
New York University Medical Center
New York, New York
1980
Part II: Chapter 10
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
With such strong evidence that magnesium deficiency-or other factors that cause subnormal magnesium levels-can lead to functional and morphologic cardiovascular abnormalities, it is surprising that there has been so little clinical application of these findings. It is to be hoped that the detailed case reports published by Chadda et al. (1973b) and Iseri et al. (1975), in which they described rapid correction by magnesium of arrhythmias that had been refractory to the widely accepted therapeutic modalities, will stimulate others to consider magnesium treatment and evaluation of the magnesium status of patients with cardiac, and especially life- threatening arrhythmias. It must be cautioned that severe hypomagnesemia is not a necessary finding. For example, Chadda et al.(1973b, 1976/1980) found only slightly subnormal serum magnesium levels, but histories of diuretic intake and myocardial infarctions (which cause magnesium loss) in patients with a high incidence of ventricular ectopia. Iseri et al. (1975) reviewed the clinical states and drugs associated with magnesium deficiency and loss, and pointed out that magnesium deficiency can clearly exist without hypomagnesemia. They cited a reference (Loeb et al., 1968) that demonstrated that hypomagnesemia can predispose to arrhythmia (which eventually responded to standard therapy without magnesium repletion). Noting the rapid response to magnesium of hypomagnesemic arrhythmias reported by others (Scheinman et al., 1969; Rosefsky, 1972; Chadda et al., 1973a) they instituted magnesium therapy in refractory arrhythmic patients after taking a blood specimen for pretreatment magnesium values, and affirmed the rapidity with which the arrhythmias were corrected.
Unfortunately, magnesium determinations are rarely part of the routine electrolyte evaluation of patients with arrhythmia. Even when detected, its correction may be delayed until failure of classic approaches; addition of magnesium results in rapid amelioration of rhythmic disturbances (R. Singh et al., 1975). Among those who have diagnosed hypomagnesemia, electrocardiographic evaluation is reported only occasionally. Thus, there are no firm data at present as to the frequency with which both abnormalities coexist. In a pilot study, Chadda et al. (1977) found that 10 among 12 patients with hypomagnesemia (7 secondary to alcoholism, 2 secondary to malabsorption and intestinal fistulae, 2 as a result of postsurgery hyperalimentation, and 1 in chronic renal failure), 10 had cardiac arrhythmias. Seven had ventricular tachycardia, fibrillation or more than 6 premature beats (VPBs) per minute, or atrial arrhythmia with hypotension. All of the patients with VPBs had a prolonged QT interval. Two patients had electrical alternans. The serious arrhythmias of 4 of the patients had been unresponsive to any treatment other than magnesium. All of the arrhythmic patients improved when magnesium was given.
When one considers the unreliability of serum magnesium as an index of the cellular magnesium status, the difficulty of correlating (occult) magnesium deficiency with ECG abnormalities or predisposing cardiomyopathies can be readily appreciated.
In this section, attention is given to the dramatic responses of arrhythmias to magnesium therapy and to the conditions in which such responses have been described. Consideration is also given to the nature of the magnesium therapy, and to the differences in results obtained when it is used simply as a pharmacologic agent, and when it is given as sustained therapy (in which event one may presume that an underlying deficit may be repaired). It is possible that prophylactic long-term use of magnesium supplements, possibly from the beginning of life, might be preventative of the cardiomyopathies and arterial lesions that predispose to arrhythmias (supra vide), as well as of some skeletal and renal disorders (infra vide).
Intravenous use of magnesium to correct arrhythmias was demonstrated by Seekles et al., (1930), who found that it was useful in reversing arrhythmia caused by calcium treatment of the convulsions and tetany of cows with "grass staggers" of early lactation. This group soon demonstrated this disorder in cows that were hypomagnesemic and showed that it developed in areas and at times when there was a high potassium/magnesium ratio in their forage (Sjollema and Seekles, 1932). In a few years this syndrome was shown to be associated with cardiovascular lesions that involved the subendocardium and the myocardium, including the Purkinje cells (Moore et al. 1936). Thus, these studies of the correction by magnesium of calcium-induced arrhythmia might have been the result of correction of calcium-intensified magnesium deficiency. More recently, Ghana and Rabah (1977) have shown that magnesium reduces the vulnerability to electrically induced ventricular premature contractions (VPC) and of ventricular fibrillation (VP) of normal intact dogs, heart-lung preparations, and digitalized dogs (Table 10-1). Intravenous magnesium chloride solution, providing 100 mg of magnesium per kg of dog, increased the millivoltage tolerated by the intact dogs by 53% and over 100%, respectively, before they developed VPCs and VF. The heart-lung preparations tolerated 72% and 130% higher millivoltages before developing the VPCs and VF. Three of the digitalized dogs did not survive the VP phase before magnesium was to be given.
{kind=link}
It is of interest that intravenous calcium, especially when given to patients with arrhythmias of digitalis toxicity, has had serious, sometimes catastrophic, effects (Lloyd, 1928; Bower and Mengel, 1936; Berliner, 1936; Golden and Brams, 1938). The potentiation of toxicity of cardiac glycosides, not only by calcium, but by other agents (e.g., catecholamines) that increase myocardial uptake of calcium suggest that potentiation of calcium influx into the myocardium by cardiotonic alkaloids (Review: Nayler, 1967) is potentially harmful. Cardiotonics simultaneously cause magnesium efflux from the myocardium (Hochrein et al., 1967; Wilke and Malorney, 1971) and inhibit magnesium-dependent cardiac mitochondrial and microsomal enzymes (Review: Seelig, 1972). Relevant to these findings is the observation that quinidine causes focal mitochondrial damage (Hiott and Howell, 1971) and that both magnesium and potassium chloride have significantly (p < 0.001) reduced cardiac necrosis caused by digitoxin (Savoie et al., 1969).
Noting the risk of using intravenous calcium in measuring circulation time, which even in noncardiac patients causes flattened or inverted T waves in 92% of the subjects, flattened or inverted P waves in 54%, and marked bradycardia in 67%, M. Bernstein and Simkins (1939a,b) contrasted the effects of magnesium as a circulation-time reagent. They investigated the electrocardiographic effects of 10 ml of 10% magnesium sulfate solution (100 mg of magnesium) in 100 patients: 66 with and 34 without cardiovascular disease. They found no deleterious effects on the heart. There were inconsistent ECG changes in 26 of the 66 cardiac patients during or after the injection that were limited to the T waves and the QRS complexes (usually increased amplitude). Comparable benign changes were seen in 10 of 34 noncardiovascular disease patients. They had undertaken the study because of the statement that had been made that "sudden death following the injection of a magnesium salt … is not an uncommon occurrence," and the demonstration (with massive doses of magnesium) that magnesium adversely affected cardiac rhythmicity (J. R. Miller and VanDellen, 1938). P. K. Smith et al. (1939) demonstrated, for example, that cardiac arrest could indeed be produced by magnesium, but not below serum magnesium levels of 27 to 44 mEq/liter. Thus, it is important to distinguish between pharmacologic doses of magnesium, such as are used in the treatment of arrhythmias, and toxic doses. Serum levels of magnesium should be kept below 5.5 mEq/ liter (Iseri et al., 1975; Iseri and Bures, 1978), which gives an ample safety margin. Only levels above 10 mEq/liter have been shown to cause toxicity (Review: Engbaek, 1952).
B. M. Cohen (1952), who reviewed digitalis toxicity and its treatment, summed up the arrhythmias produced (nodal and paroxysmal tachycardias, ventricular extrasystoles often producing bigeminy or trigeminy, and heart block) and mentioned contraindications of digitalis therapy, including paroxysmal ventricular tachycardia, and coronary insufficiency without cardiac failure. He also cited the risk of calcium therapy in digitalized patients and the additive toxic effects of digitalis and catecholamines. It is noteworthy that the arrhythmias described are also seen in magnesium deficiency and that magnesium deficiency or loss increases susceptibility to digitalis toxicity in animal and man (Vitale et al., 1961, 1963; Kleiger et al., 1966; Caddell, 1967; Wacker and Parisi, 1968; Ono, 1971/1973). Furthermore, patients with digitalis toxicity not infrequently have subnormal magnesium levels (Kim et al., 1961; Beller et al., 1974; R. Singh et al., 1976).
Magnesium's antiarrhythmic effects were first demonstrated in man in digitalis toxicity (Zwillinger, 1935). This effect has also been demonstrated experimentally (Zwillinger, 1935; Szekely, 1946; J. Stanbury and Farah, 1950; Szekely and Wynne, 1951; Gendenshtein and Karskaya, 1963; Bajusz et al., 1969; Seller et al., 1970a,b; Neff et al., 1972; Specter et al., 1975) and affirmed in man (Boyd and Scherf, 1943; Szekely, 1946; Zimdahl, 1946; Freundlich, 1946; Szekely and Wynne, 1951; R. Par sons et al., 1959; Michel, 1966; Kabelitz, 1968; Condorelli, 1971/1973; Lossnitzer, 1971a,b; Rotman, 1971; Iseri et al., 1975; R. Singh et al., 1976; Iseri and Bures, 1978). The long time lag between the first cluster of clinical reports and the more recent observations on magnesium's efficacy in digitalis arrhythmia and in other arrhythmias is probably a consequence of its early use only as a pharmacologic agent that had transient activity and occasionally caused increased irregularity of rhythm (B. M. Cohen, 1952). Since then, the substantial evidence that loss of magnesium from the myocardium can cause cardiomyopathies that predispose to arrhythmias justifies reexamination of how best to utilize magnesium in their treatment.
{kind=link}
Electrocardiographic changes caused by coronary ligation in dogs have responded to intravenous infusion of magnesium salts. Harris et al. (1953) showed that the duration of ischemic tachycardia and ectopic rhythm was shortened in 46% of the dogs infused with magnesium as the sulfate and in 70% of the dogs infused with magnesium as the chloride, at a dose of 1 mEq/liter. Clark and Cummings (1956) found that each of three successive MgSO4 infusions corrected the ischemic tachycardia and multifocal ventricular arrhythmia (J. R. Cummings, personal communication). Locke-Ringer solution lacking magnesium did not influence ischemic fibrillation, but when either 0.05 mEq or 2.0 mEq of magnesium was added-either to Ringer's solution or to 0.9% saline-there was protection against fibrillation. Dogs with persistent ventricular tachycardia and ectopic extrasystoles (after two-stage coronary ligation) responded to repeated injections (up to seven) of MgNa2EDTA solution (50-100 mg/kg body weight) by a 25% decrease in heart rate, and sometimes by transitory restoration of the sinus rhythm on the day after the ligation. The effect of the infusions were sustained somewhat longer, but were still transient, two days after the ligation (Gendenshtein and Karskaya, 1963). The aspartate salts of magnesium and potassium, in combination, were protective against ischemic ECG changes in rabbits with coronary arterial ligation (Weber et al., 1958) and against ECG of asphyxia in guinea pigs (Hochrein and Lossnitzer, 1969)
Isolated hearts, under hypoxic conditions, have shown less reduction of systolic amplitude and other ECG changes of anoxia when suspended in fluids containing magnesium and potassium aspartates; chloride salts of the cations were less effective (Laborit et al., 1957; Weber et al., 1958; Trzebski and Lewartowski, 1959: LaMarche and Tapin, 1961; LaMarche et al., 1962; H. Rosen et al., 1964: LaMarche and Royer, 1965). Some of the benefit might reflect the coronary vasodilation shown to be produced by magnesium and potassium sulfate or chloride (Elek and Katz, 1942; Scott et al., 1961; Review: Haddy and Seelig, 1976/1979). The aspartate salts were more effective than the chlorides in the in vitro studies.
10.1.2 .2. Magnesium in Clinical Arrhythmias of Ischemic and Unknown Origin
Having demonstrated in vitro that magnesium sulfate has coronary vasodilator activity, Elek and Katz (1942) recommended its use as a pharmacologic agent in paroxysmal tachycardia associated with myocardial ischemia. Boyd and Scherf (1943) corrected paroxysmal auricular tachycardia (PAT) by giving 10-15 ml of 15% MgSO4 or 10 ml of a 30% solution intravenously (in 10 of 19 treatments). Comparable dosage was effective in 9 of 13 patients with PAT and in 1 with Wolff-Parkinson-White syndrome (Szekely, 1946), restoring sinus rhythm and decreasing the heart rate. The latter investigator noted that the patients most responsive to magnesium therapy were those who had advanced heart disease with congestive failure. One may speculate that such patients are likely to have received long-term diuretic and cardiotonic therapy, and thus to be most magnesium depleted. Neither group found any effect of magnesium on auricular flutter or fibrillation.
Despite these early promising results, and the experience of clinicians from the British Commonwealth (England, South Africa, Australia, and England: Malkiel Shapiro et al., 1956; 1960; Malkiel-Shapiro, 1958; R. S. Parsons, 1958; Parsons et al., 1959, 1960, 1961; Agranat, 1958; Marais, 1958; Teeger, 1958; Anstall et al., 1959; Browne, 1961, 1963, 1964a,b; Hughes and Tonks, 1965; Tonks, 1966) with the efficacy of long-term treatment of patients with acute or chronic IHD (rationale based on magnesium's effects on blood coagulation and lipids), the clinical use of magnesium in cardiovascular disease has been slow to gain acceptance in America. It has been utilized, usually with potassium, with and without heparin, and as organic salts (e.g., nicotinate and aspartate) parenterally (with and without glucose and insulin) immediately after infarction, and orally in the management of postinfarction patients and those with angina pectoris. Such use has been described in studies from the European continent (Hoffman and Siegel, 1952; Laborit et al., 1958; Melon, 1960; Thurnherr and Koch, 1962; Larcan, 1963; Perlya, 1965; Kanther, 1966; Kenter and Falkenhahn, 1966; Rigo et al., 1965; Köhler, 1966; Laborit, 1966; Larcan, 1966; Maté et al., 1966; Michel, 1966; Nieper and Blumberger, 1966; Pillen, 1966; Stepantschitz and Fröhlich, 1967; Savenkov et al., 1971). Most of the reports have been uncontrolled clinical trials, sometimes large series of cases that were compared with prior series treated identically except for the magnesium (and potassium) salts. (Representative treatment regimens are entered on Table 10-3 and Table 10-3 continued.)
{kind=link}
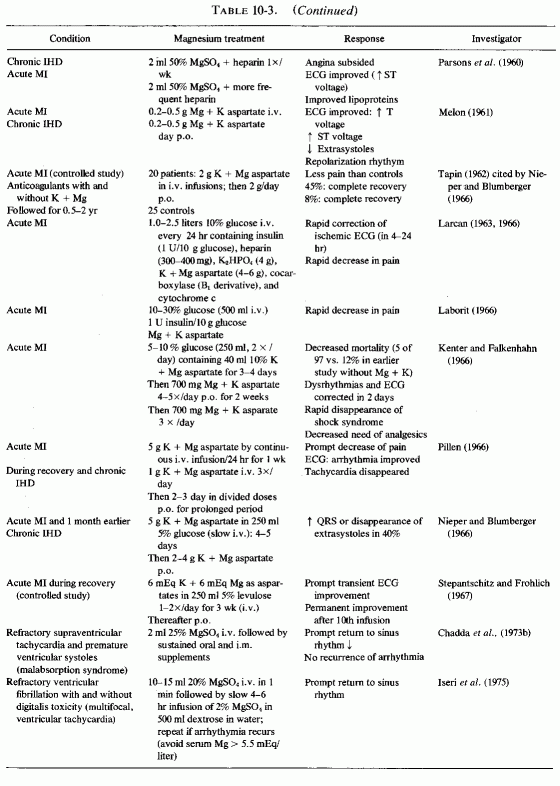{kind=link}
Whether the combination of magnesium and potassium aspartate salts, given in high doses for treatment of the acute infarction and then followed by prolonged oral therapy for indefinite periods, provides better results than does the inorganic sulfate, which was somewhat similarly used only in the South African and Australian studies, cannot be averred. The studies evaluated different parameters; comparably better results were obtained with prolonged than with short-term therapy. Nieper and Blumberger (1966) refer to a controlled study with the mixture of magnesium and potassium aspartates in 45 patients with acute myocardial infarction (Tapin, 1962). Classical anticoagulant and supportive therapy was provided the 25 control patients; 2 g of magnesium and potassium aspartate were added to the daily infusions of the 20 patients in the test group until infusions were discontinued, at which time the patients were given the same daily dosage orally. [Nieper and Blumberger (1966) commented that their own experience indicates that 5 g daily in 250 mg of 5% glucose, given by slow intravenous infusion, for 4 to 5 days is preferable, to be followed by 2 to 4 g daily orally thereafter.) Nonetheless, Tapin (1962) found that, even with the low dosage used, his magnesium and potassium aspartate treatment group had a somewhat lower death rate in the hospital (40% versus 56% among those on standard therapy). The real difference was manifest among the survivors (6 months to 2 years follow-up). The magnesium and potassium aspartate treated group showed complete recovery in 45%; only 8% of the controls recovered completely. Pillen (1966) and Nieper and Blumberger (1966), using the higher dosage regimen routinely in their acute-infarct patients, found good to excellent results in 16 of 19 patients, and recommend immediate intravenous administration of magnesium and potassium aspartate as part of the emergency treatment, even before the patient reaches the hospital. They found rapid improvement of the ischemic ECG (within 12-24 hours), as well as rapid decrease of pain, most patients requiring no analgesic therapy.
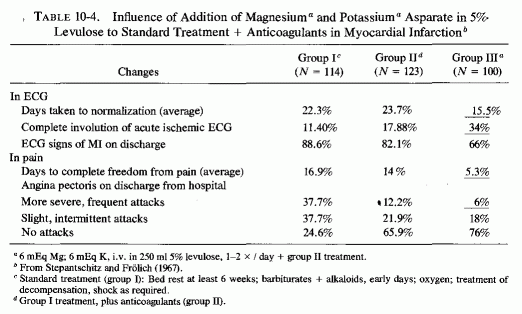
Stepantschitz and Frohlich (1967) compared the outcomes in three groups of patients hospitalized for six weeks after an acute myocardial infarction: Group I (114 patients) received standard supportive therapy that included oxygen, sedation, alkaloids, treatment of shock with corticosteroid and epinephrine, and antiarrhythmia agents as necessary. Group 11(123 patients) were also given anticoagulants. Group III (100 patients) were given magnesium and potassium aspartate in addition to the therapeutic regimen given group II. Each patient in group Ill was given 6 mEq of magnesium and potassium and 12 mEq of aspartate in 250 ml of 5% levulose once or twice daily for three weeks, and the same dosage for the remaining three-week period of hospitalization. The death rate in group I was 46.5%, versus 13 and 15% in groups II and III. However, in noting the comparable mortalities in the groups receiving anticoagulants (II) and magnesium and potassium aspartate (III), the authors noted that the patients in group III had had 13 times as many recurrent infarctions as had the other two groups, and thus had the poorest prognosis. They tabulated the criteria for the effects of treatment(Table 10-4) and pointed out that the most striking advantage of the magnesium and potassium aspartate therapy was in the time taken to achieve complete freedom from pain (average of 5.3 days, versus 16.8 and 14 days in groups I and II0. The average time taken for the ECG to return to near normal was 15.5 days in group III, versus 22.3 and 23.7 days in groups I and II. Complete involution of the ECG signs of infarction occurred in 34% of the patients in group III, and in 11.4% and 17.9% of those in groups I and II.
{kind=link}
Using the transcardiac iontophoretic method of giving magnesium to patients with myocardial infarction, Köhler (1960) found complete to almost complete relief of pain in 88 of 100 patients, and marked improvement in 12, as compared with complete relief in none, and marked to almost complete relief in only 27 who received placebo iontophoresis. In the remaining placebo group, 34 were unchanged or worse and 36 showed only slight improvement. He later commented (Kohler, 1966) that the iontophoretic procedure carries in only the cation. Kucher (1966), using the same procedure, but with both magnesium and potassium salts, reported that 180 to 184 patients (classified as angina pectoris with myocardial degeneration) improved, and all 22 patients who had recent infarctions improved. Among those who had refractory auricular fibrillation, extrasystoles, or paroxysmal tachycardia, 9 of 16 improved.
10.1.2.3. Glucose Solutions and Insulin to Increase Myocardial Magnesium and Potassium Uptake
Laborit (1958) considered hypertonic glucose solutions useful in attaining a normal myocardial electrolyte gradient, (for repolarization) and recommended the use of aspartate salts of Mg + K. Sodi-Pallares et al., (1962, 1966, 1979) suggest the addition of insulin to reverse the ECG signs of ischemia. Kones (1975) has evaluated the clinical response of patients with infarction and reported that glucose-insulin- potassium therapy is a useful therapeutic adjunct. Opie and Owen (1976) have provided experimental evidence that such treatment increases the arteriovenous coronary difference of glucose, decreases the free fatty acids, accelerates the fall of the epicardial ST segment, and prevents the small rise in the ST segment in the peri-infarct and nonischemic zones. Gavrilescu et al. (1974) have shown that slow (over 1-hour period) i.v. infusion of 3 g of potassium and magnesium asparate in 200 ml of physiological saline lowers the elevated levels of free fatty acids that develop during the first hour after an acute myocardial infarction (p. 225).These findings support the contention that such treatment has beneficial effects on tissue metabolic, histologic, and electrocardiographic criteria of ischemic damage. In commenting on Sodi-Pallares' and Opie's findings in the clinical and experimental situation, and Sodi-Pallares' (1976) reminder that diuretics and antiarrhythmic therapy are contraindicated with the polarizing treatment, Bing (1976a, b) observed that the metabolic findings with this form of therapy might well provide a piece of the Rosetta stone. He indicated, however, that until the etiology of ischemic heart disease can better be defined, it will continue to be difficult to bridge the gap between fundamental and applied knowledge.
The data presented in this volume provide considerable evidence that cellular magnesium deficiency can be another key to the etiology of ischemic heart disease and other cardiomyopathies. Since administration of insulin and glucose has been shown to accelerate the uptake of 28Mg by the heart more than twofold (Aikawa, 1960a), and the magnesium ion seems to be essential for maintaining tissue response to insulin (G. Bhattacharya, 1961), addition of magnesium to the polarizing solution would seem advisable. It is provocative that Bajusz (1964, 1965b) found that the partially protective effects of either magnesium and potassium chlorides or aspartates were markedly increased by simultaneous administration of glucose and insulin. Another justification for including magnesium in the polarizing solution is its requirement for the enzyme systems necessary for accumulation of potassium against a concentration gradient (Review; Seelig, 1972).
A metabolic approach to the treatment of endomyofibrosis of the adult (with abnormalities of the ST segment and Q wave) incorporated magnesium and vitamin B1 as well as insulin, glucose, and potassium, to enhance glycolytic metabolism (Michon et al., 1959). Larcan (1966) later reiterated the value of this approach, using cocarboxylase (a B1 metabolite) instead of thiamine in the treatment of patients with myocardial infarction. He stressed the importance of including magnesium. He reproduced representative ECGs from representative cases from his series of 40 cases, and commented that most striking was the much more rapid analgesic effect in the metabolically treated patients than in a control group that was treated by bed rest, anticoagulants, and opiates. Asthenia was also notably diminished, and the ischemic ECG changes regressed rapidly, the improvement beginning as early as four hours after the first ECG on hospitalization, and being definitive by the end of the first to second day of the infusions.
10.1.2.4. The Role of the Anion
In most of the clinical trials, magnesium sulfate has been the salt used, and in the United States it is the only readily available parenteral preparation. Ischemic arrhythmia in dogs responded somewhat better to magnesium chloride than to magnesium sulfate at a 1 mEq/liter dose of magnesium (A. Harris et al., 1953). However, the numbers were too small for significance to be determined. Selye (1958d,) showed that not only phosphate, but also sulfate, sensitizes the heart to cardiopathic agents, whereas the chloride (of magnesium or potassium) is protective. He found no superiority of the aspartate or orotate salts of magnesium and potassium to the chloride salts as cardioprotective agents in his experimental models (Selye, 1958g). More recently a hydrochloride salt of magnesium and potassium aspartate has been investigated, and found to be better absorbed and utilized, and to be more effective than the aspartate salts in experimental cardiomyopathic models (Classen et al., 1973, 1975, 1976, 1978; Ebel et al., 1975). Neither magnesium sulfate nor magnesium aspartate were effective against cardiac necrosis induced by epinephrine plus a mineralocorticoid, whereas magnesium chloride and magnesium aspartate hydrochloride each exerted significant protective effects (Classen et al., 1975, 1978). These investigators concluded that it is necessary to correct, not only the magnesium deficit, but the hypochloremic alkalosis in metabolic myocardial necrosis. Lehr et al. (1972) concur that it is necessary to provide both magnesium and chloride to protect against experimental myocardial necrosis of widely different natures (Lehr, 1965, 1969; Lehr et al., 1966).
10.2. Formulation of a Metabolic Therapeutic Program for TreatingCardiomyopathies and Arrhythmias
It is important to consider all of the positive and negative findings from animal and human studies in determining a safe, effective approach to the treatment of cardiomyopathic disease, whether of ischemic or other origin. Because magnesium deficiency or loss from the myocardium has been repeatedly implicated in experimental cardiomyopathy, and because magnesium is cardioprotective, it should be included in treatment programs, such as in the polarizing treatment. Sodi-Pallares (1969) cautions against the use of diuretics and corticoids (which cause loss of magnesium, as well as of potassium) and such inotropic drugs as digitalis, quinidine, and catecholamines, unless there is pulmonary edema or atrial fibrillation and ventricular tachycardia. Since inotropic drugs and some diuretics (e.g., thiazides) increase calcium retention and, in the case of the glycosides and catecholamines, increase myocardial calcium uptake and lipolysis, caution should also be exercised in treating hypocalcemia of cardiac patients with intravenous calcium salts. Potassium chloride is readily available and should certainly be included in the therapeutic regimen. (The author suggests that it be used with magnesium in a polarizing solution incorporating dextrose, water, and insulin.) Unfortunately, in the United States, magnesium is available for parenteral use more readily as the sulfate than as the chloride. Perhaps the aspartate-HCl salt of magnesium will become available in the United States, as it is in Europe.
Table 10-3 indicates the therapeutic regimens that have been effective in the treatment of the acute ischemic event and in hypomagnesemic arrhythmia. In open-heart surgery, magnesium has been a useful additive to the pump-prime (optimum concentration to be proved, supra vide) and has been used as an intravenous bolus (0.1 g/kg) to facilitate postoperative defibrillation (Buky, 1970). Magnesium chloride (100 mg Mg) has also been recommended, pre- and postoperatively, to prevent arrhythmias (Khan et al., 1973; Holden, 1978). The emergency therapeutic dosage of magnesium, as described by Iseri et al. (1975) is recommended, with the modification that after the bolus of magnesium, the maintained infusion should be 5-10% dextrose in water plus insulin (0.1 unit/g dextrose), and potassium (3-6 mEq) and magnesium (3-6 mEq) as the chloride or aspartate hydrochloride, if available. Possibly, the water-soluble B vitamins and vitamin C should be added to the infusion in "stress-formula" concentrations. Investigations are required to determine the optimal formulation.
---------------------------------
Part III: Chapter 11
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
Relatively little attention has been paid to the importance of magnesium in bone metabolism, except to the degree that it affects the activity of the parathyroid glands and C cells and their secretion of parathyroid hormone PTH and calcitonin (CT), and the response of target organs. However, experimental magnesium deficiency causes abnormalities in skeletal structure, enzymes, and mineralization that resemble some of those seen in several clinical bone diseases. Depending on the degree and duration of the magnesium deficiency and concomitant dietary or iatrogenic imbalances (of magnesium with calcium, phosphates, vitamin D, and other calcemic agents), the pathologic skeletal findings can range from osteopenias to osteosclerosis. The effects of vitamin D, calcium, and phosphorus on magnesium requirements and on skeletal responses have been intensively studied, particularly in the 1930s, when vitamin D toxicity was the focus of much attention. Many of the results are conflicting, probably due to the dietary variations, and to species differences in requirements (i.e., of vitamin D). Only those portions of the PTH/CT/Mg data that deal directly with magnesium and bone are considered here. Much of that relating to gestational abnormalities has already been discussed. The relatively little information found on heteroionic magnesium/calcium exchange in bone, and on the magnesium interrelationships between the phosphatases that affect mineralization, alkaline phosphatase and pyrophosphatase, are brought into focus as possibly providing some insight into the conflicting and confusing data on mechanisms of pathologic skeletal processes.
Largely disregarded in the treatment of bone disease is the possibility that some of the therapeutic agents (used to increase bone mineralization) might adversely affect bone metabolism by causing loss of skeletal magnesium. Calcium, phosphorus, and vitamin D all increase magnesium requirements; the intakes of all have been rising during this century, while that of magnesium has been falling. Since plasma levels of magnesium are maintained within very narrow limits, even in the face of insufficient intakes or excessive losses, the magnesium is mobilized from the tissue stores. Bone constitutes the largest total source; it contains two-thirds of the total body magnesium (Review: Heaton, 1971). Much of bone magnesium is quite labile, especially in young animals. Were the bone magnesium merely an inert storage depot, this would be a benign means of providing magnesium for the function and structure of life-preserving tissues (e.g., cardiovascular and renal), as well as preventing acute neuromuscular signs of magnesium depletion. For short periods of time, and more in young than in older individuals, availability of bone magnesium probably serves as a safety device that prevents serious systemic signs of magnesium deficiency. However, long-term loss of magnesium from the bone causes disturbances of bone modeling, remodeling, and turnover, with resultant bone abnormalities. Depending upon the supply of the calcemic agents or phosphate, it can give rise to formation of brittle chalky bones or to osteopenia. The mobilized bone constituents contribute to the renal damage of magnesium deficiency.
Because the amount of magnesium bone is only 1/40 to 1/50 that of calcium (Duck- worth et al., 1940), relatively few investigators have given it much consideration as a significant bone mineral, either in bone metabolism or as a source of emergency magnesium supply. Bone magnesium is an important source, especially in young animals (McAleese et al., 1961), an observation supported by the drop in bone magnesium immediately after convulsions of magnesium deficiency (Orent et al., 1934; Martindale and Heaton, 1964). Differences in responses to vitamin D, PTH, and CT influence the mobilization of magnesium during magnesium deficiency and have led to diverse findings. Many of the studies have dealt with the influence of magnesium deficiency and repletion, with high and low calcium, phosphorus, and vitamin D intakes, on metabolic balance. They are not considered here, unless bone values are also given, since positive balances (e.g., of calcium and phosphorus) can be achieved by metastatic calcification, as well as by increased bone mineralization and can occur even with bone demineralization. Also, failure to exhibit negative magnesium balance under conditions that cause abnormal bone structure might be related to the initial shift of bone magnesium and calcium (e.g., the increase in bone magnesium/calcium ratio in rickets).
Some of the disparate findings in the different studies might well be the result of use of widely differing diets in the magnesium deficiency studies: diets that provide 3200 to 8000 parts per million (ppm) of calcium, 1900 to 5100 ppm of phosphorus, and 1150 to 1,000,000 IU of vitamin D per kilogram of diet mix, and 3 to 100 ppm of magnesium (Larvor and Durlach, 1971a). In some of the studies analyzed and tabulated by Larvor and Durlach (1971a), only the magnesium provided was indicated. Thus, the studies cited in the following sections are not strictly comparable.
Most studies of hypervitaminosis D are in rats, which are commonly fed rations rich in calcium and phosphate, as well as in vitamin D. All three of these supplements cause magnesium loss (Reviews: Heaton, 1971; Larvor and Durlach, 1971b; Seelig, 1971). High calcium intakes compete with magnesium for intestinal absorption and renal tubular reabsorption (cited reviews), and high calcium extracellular levels result in exchange of bone magnesium for calcium.
Orent et al. (1934) were the first to note that rats on a low-magnesium, very high-calcium diet (Mg/Ca= 5/ >3000), also fed vitamin D lost about half the original percentage ash magnesium, but doubled the percentage ash calcium in their long bones. The magnesium was 1/3 normal for the same-age control rats. They noted that in rats sacrificed during convulsions, the magnesium level rose in the blood and dropped sharply in the bones, suggesting rapid mobilization of bone magnesium at that time. Nonetheless, the total accretion of bone magnesium exceeded the amount fed, and the authors speculated that it might have derived from organs such as liver, kidney, and heart, and possibly from muscle, organs which also increased in calcium content. They suggested that lowering of the skeletal magnesium ratio might have been caused by their having added vitamin D to the rats' rations. Comparable reduction in bone magnesium was reported by Cunningham (1936b) in rats fed the same magnesium-deficient diet (Kruse et al., 1932). Watchorn and McCance (1937) provided cod liver oil rather than viosterol to the rats that they maintained on a subacute magnesium-deficient regimen for up to three months. Notable were renal calcification and hepatic and skeletal damage. The long bones and teeth were brittle, and the teeth were loose in their sockets. Even though few of the many studies of vitamin D toxicity (which emphasized renal and cardiovascular damage) provided magnesium values, some of the findings (which subsequent work suggests might have been contributed to by magnesium depletion caused by the regimens) are included here. For example, rats developed overcalcification of bones and teeth (which is suggestive of a process that inhibits mobilization of bone minerals) when they were given high-dosage vitamin D, as well as diets rich in calcium and phosphorus (L. J. Harris, 1932; Shelling and Asher, 1932). In the late stage of moderate hypervitaminosis D, or with very high doses, there were cessation of osteogenesis and bands of less calcified bone near the epiphyses. (The histological changes described are much like those reported in magnesium-deficient rats and in human osteopetrosis.) Storey (1960) noted that intermittent hypervitaminosis D produces similar lesions. Comparable hypercalcification of bones, which lost 74% of control magnesium content, was found in magnesium-deficient chicks supplemented with calcium and vitamin D (C. Reddy et al., 1973). They also had increased unmineralized osteoid and cortical thickening, that was reversed rapidly on magnesium repletion. A recent study with hypervitaminosis D in pigs clarified the nature of the bone pathology with increasing doses. At 5 and 25 times the recommended dose there was osteopetrosis; at higher doses there were hypercalcemia and hypophosphatasia (Chineme et al., 1976).
On the other hand, it was suggested that rats that developed hypomagnesemia during their overdosage with vitamin D, and that did not exhibit hypermagnesuria, might be depositing magnesium in their bones (Richardson and Welt, 1965). Wallach et al. (1966) confirmed this premise in dogs on 1% dietary calcium intake, given very high vitamin D doses, that became hypercalcemic and hypomagnesemic. Their bones had only slightly increased total calcium and moderately increased (p < 0.2) exchangeable calcium. Their total bone magnesium, however, had increased significantly (p < 0.001), but there was little change in the exchangeable magnesium content.
The total bone mineral distribution of the dogs given short-term toxic doses of vitamin D (Wallach et al., 1966) resembles that reported in the early rickets studies in rats [Malcolm, 1904; Mellanby, 1926 (cited by McHargue and Roy, 1930)]. Since these animals were hypomagnesemic, as were rats overdosed with vitamin D (Hanna, 1961a; Harrison and Harrison, 1964), it can be speculated that they were in the early stage of development of vitamin-D-resistant rickets (i.e., hypervitaminosis D rickets: Ham and Lewis, 1934). Longer-term hypervitaminosis D plus high calcium intakes, as in the Watchorn and McCance (1937) and Storey (1960) studies, might be experimental models of infantile hypercalcemia, which is associated with osteosclerosis as well as with metastatic calcification (Review: Seelig, 1969b).
Despite the magnesium loss caused by the vitamin D and calcium excesses, caution must be exercised in repleting the magnesium. Whittier and Freeman (1971) have demonstrated that metastatic calcification has been potentiated by giving magnesium to rats with hypercalcemia caused by hypervitaminosis D. This recalls the speculation that the use of magnesium laxatives, to manage the obstipation of hypercalcemic children, might have contributed to their metastatic calcification (Creery, 1953; Lowe et al., 1954; Review: Forfar thesis). The rationale for this paradoxical observation is considered elsewhere in this chapter. It is important to keep in mind now that hypophosphatemic rickets, refractory to high dosage vitamin D and calcium, has been reported to be responsive to magnesium.
Fetal and neonatal spontaneous fractures and lesions resembling those of osteogenesis imperfecta and hypophosphatasia develop in pups of rats given high doses of vitamin D and in infants born with intrauterine growth retardation, both conditions that might be related to fetal magnesium deficiency.
Early or acute magnesium deficiency has been shown to stimulate PTH secretion, but the concomitant hypercalcemia in the experimental model and most clinical conditions in which hypervitaminosis D plus high calcium intakes play a role would function to decrease PTH secretion, outweighing the stimulant effect of magnesium deficiency. Additionally, early and acute magnesium deficiency has stimulated CT secretion, an effect enhanced by hypercalcemia (Stachura and Pearse, 1970). Thus, the overall effect on bone of diets low in magnesium and high in calcemic agents is decreased mobilization of bone calcium, with replacement of surface bone magnesium by calcium.
Rats on normal diets given high-dosage vitamin D without calcium supplements or low-calcium diets were shown, in early studies, to exhibit resorption of compact bone, an effect attributed to vitamin-D-induced bone mineral mobilization (Duguid. 1930a; L. J. Harris, 1932; Shelling and Asher, 1932). In a 1953 review, Nicolaysen and Eeg-Larsen reported that the dominant feature of hypervitaminosis D is dissolution of formed bone and dense calcification of hypertrophic cartilage.
Duckworth et al. (1940), whose magnesium-deficient rats had much less bone calcification than did those of Orent et al. (1934), did not list vitamin D as a dietary constituent. They found that weanling rats, kept on a diet adequate in calcium but low enough in magnesium to result in tetany or convulsions and death by 6 days to a month, had less growth and markedly less magnesium (percent in ash) in their bones than did littermates on the same diet but supplemented with magnesium. In contrast, the magnesium-deficient rats had no decrease (percent in ash) of calcium or phosphorus. In fact, they had a slightly increased percentage of bone ash calcium. Those on the deficient diet for 16 and 23 days exhibited the greatest percentage loss of magnesium as compared to adequately pair-fed rats (0.39 → 0.34% versus 0.83 → 0.74% Mg in bone ash). Rapid replenishment of the bone magnesium was exhibited by rats fed deficient diets for 6 days and then adequate diets for 10 days. The bones of the rats that survived the magnesium-deficient period had more fragile bones than did those reared on adequate rations, and give histologic evidence of abnormal matrix. They then found that rats fed diets deficient in both calcium and magnesium survived longer than did those fed diets adequate in calcium but low in magnesium (Duckworth and Godden, 1941). The rats low in both cations more quickly mobilized more magnesium from their bones, a possible explanation of their longer survival. The rate of bone growth determined the amount of the magnesium that could be liberated because of the demand of the skeleton itself for magnesium. They then showed that when the diet was free of calcium but contained no less than 6 ppm of magnesium, the demineralized bone ash contained progressively more magnesium and less calcium (Duckworth and Godden, 1943). Thus, to a limited extent, the magnesium replaced calcium in the bone crystal. This did not occur with deficiency of both cations.
The mobilization of bone mineral (particularly calcium, the major bone mineral, but also magnesium) by excess vitamin D with low calcium and magnesium intakes or body reserves might be a direct effect, as has been shown with vitamin D metabolites (Trammel et al., 1969; Raisz et al., 1972; Reviews: Norman and Henry, 1974; Norman et al., 1975/1977; DeLuca, 1976) or one that is mediated by secondary hyperparathyroidism. That hypocalcemia causes increased PTH secretion is well established. The effect of hypomagnesemia is neither as well known nor as clear-cut. Larvor et al. (1964a) demonstrated that magnesium deficiency (in a calf on normal calcium and vitamin D intakes) caused hyperplasia and osteitis fibrosa. Indirect evidence of increased PTH secretion in rats on diets low in magnesium but adequate in calcium was provided by investigators who prevented hypercalcemia in magnesium-deficient rats by parathyroidectomy (Kukolj et al., 1965; Gitelman et al., 1965, 1968b). I. Clark (1969b) provided evidence that magnesium deficiency in rats fed adequate calcium and phosphate exerts a slight stimulant effect on PTH secretion.
In vitro studies have provided direct evidence of the PTH secretory effect of magnesium deficiency. Perfusion of the parathyroids of goats and sheep (which are separate from their thyroids), with hypomagnesemic, normocalcemic solution resulted in increased PTH secretion (Care et al., 1966; Buckle et al., 1968), an effect that was verified by Sherwood (1970) and his colleagues (Sherwood et al., 1970, 1972; Targovnik et al., 1971). Despite this clear laboratory evidence, severe clinical magnesium deficiency has been shown to cause relative parathyroid failure (Muldowney et al., 1970; Anast et al., 1972, 1976; Anast, 1977; Suh et al., 1971, 1973; L. Chase et al., 1974; Avioli, 1978), an effect that can be mediated by decreased PTH release (Anast, 1977) or skeletal unresponsiveness (Estep et al., 1969; C. Reddy et al., 1973; Levi et al., 1974; Medalle et al., 1973, 1976). However mediated, Forbes and Parker (1976/1980) have shown diminished bone resorption (as measured by 45 levels) in magnesium-deficient young rats.
Why a condition associated with increased PTH secretion (that mobilizes bone minerals and leads ultimately to magnesium loss, as well as hypercalcemia) should be associated with increased levels of bone magnesium in the acute studies, is difficult to explain. It is conceivable that the enhancement by PTH of mitochondrial uptake of magnesium (Rassmussen et al., 1964) might be contributory. The increase in bone magnesium, associated with hypervitaminosis D, might be correlated with a possible PTH-mediated early bone uptake of magnesium. Since magnesium participates in osteoblastic activity and osteoid formation, the net result of the imbalance produced by concomitant hypervitaminosis D and low calcium intake (and that causes hypomagnesemia) might well be the high magnesium/calcium bone ratio, and the relative excess of osteoid, such as is seen in clinical rickets and in hyperparathyroidism. It might also include the osteomalacia of malabsorption syndromes and vitamin-D-resistant rickets following high-dosage calcemic therapy.
Possibly the initial response to hypomagnesemia of the CT producing C cells is increased secretion, even in the absence of hypercalcemia (Rojo-Ortega et al., 1971). It is conceivable that this response functions to inhibit release of bone magnesium, as well as to partially counteract the mobilization of bone calcium of animals loaded with vitamin D. However, compensatory CT secretion is insufficient to counteract calcium mobilization from bones of rats given very high doses of vitamin D (Mittlemanet al., 1967).
Despite the (possible) increase in CT secretion, hypervitaminosis D (usually in adults whose calcium intake is not high) has caused hypercalcemia and bone demineralization, as well as metastatic calcification.
11.3.1. Effects on Bone Magnesium
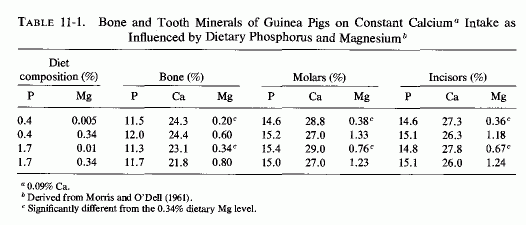
E. R. Morris and O'Dell and their colleagues studied the influence of increasing the phosphate intake on skeletal and dental structures of guinea pigs on low to normal magnesium intakes, keeping the calcium intake adequate and constant (O'Dell et al., 1960; E. R. Morriss and O'Dell, 1961). Although the calcium and phosphorus levels of the hard structures remained essentially the same in magnesium-deficient and control animals, regardless of the intake of phosphate, the animals on a diet low in both magnesium and phosphate had one-third as much magnesium in their bones and teeth as did animals on control magnesium intakes, also low in phosphate. Increasing the phosphate increased the magnesium requirements for survival, and induced changes in bone and tooth minerals (O'Dell et al., 1960). At both the low- and high-phosphate intakes, increasing the magnesium levels 70- and 35-fold, respectively, significantly increased the magnesium levels of the hard structures (Table 11-1)and prevented their structural defects. The investigators speculated that the phosphate-induced loss of skeletal magnesium caused abnormalities in the matrix. Forbes (1961) evaluated the effects of varying dietary ratios of calcium, magnesium, and phosphorus in weanling rats. He demonstrated that on marginal magnesium intakes, overt magnesium deficiency was produced only when excesses of both calcium and phosphorus were provided. The percentage of magnesium in femur ash was lowest in magnesium-deficient rats supplemented with both calcium and phosphorus and was almost as low when supplemented only with phosphorus (Forbes, 1963).
{kind=link}
In studies of the effects of magnesium depletion and repletion on rats depleted of or provided adequate calcium and phosphorus, I. Clark (1966, 1968, 1969a, b, 1971/1973, 1977) showed that the amount of each ion required or tolerated is influenced by the intakes of the others (Fig. 11-1). He also showed that femoral weight and calcification is depressed without optimal magnesium intake. In a study of bone minerals in rats on constant calcium and phosphate intakes, but on low-to-high magnesium supplements, Clark and Bélanger (1967) found declining bone calcium and magnesium as the dietary magnesium-to-calcium ratio declined. Meyer and Busse (1976/1980) reported that changing vitamin D intakes did not alter bone-magnesium levels in rats on high phosphorus intakes, although they confirmed that vitamin D slightly lowered blood levels of magnesium. They found that the magnesium-bone content of rats fed diets with slightly higher phosphorus than calcium content was slightly higher than that of rats fed diets with three times as much calcium as phosphorus. In sheep, there was also more magnesium in bone ash than when the dietary calcium to phosphorus ratio was low than when it was high.
{kind=link}
11.3.2.1. Bone Wasting
In view of the cited evidence that excess phosphate decreases bone magnesium levels, and the importance of magnesium in maintaining normal bone metabolism, the evidence that experimental high phosphorus/calcium ratios causes bone wasting is relevant. Data referable specifically to the renal calcinosis produced by diets with high phosphorus/magnesium ratios, or that cause increased phosphorus mobilization, will be discussed in Chapter 13..
Shelling and Asher (1932) studied the influence of different proportions of dietary calcium and phosphorus on bone and soft tissue calcification of rats given no vitamin D, or given moderately high to very high doses. Young rats on high P/Ca dietary ratios developed osteoporosis, which was intensified by increasing the phosphorus intake further, and further worsened by addition of large doses of vitamin D. Microscopic studies of young rats on low calcium/high phosphorus and vitamin D (40,000 times the antirachitic dose) showed a progressive decrease in the number of trabeculae with the duration of the experiment. At the end (by the 26th day), the trabeculae had been replaced by remnants of osteoid, osteoblasts, and tiny fragments of calcified material. The similarity of these abnormalities to those seen in the genetic abnormality, hypophosphatasia, and the low alkaline phosphatase levels of infants with hyperreactivity to vitamin D deserves consideration.
More recently, the risk of bone wasting (caused by high P/Ca in the diet) has been studied by Krooket al. (1971, 1975). They demonstrated nutritional osteoporosis in dogs, horses, pigs, and monkeys kept on diets with high phosphorus/calcium ratios for prolonged periods of time. The disease is characterized by hypercalcemia and hypophosphatasia; the bone damage, both in long bone and in mandibles, is related to secondary hyperparathyroidism (attributed both to the low dietary calcium and high dietary phosphate). The histological changes resemble those of osteoporosis, and are characterized by loss of matrix and by demineralization, particularly in the subperiosteal areas of the compact bone. In trabecular bone, osteocytic osteolysis occurs in the center, and the trabeculae become thinner. It is likely that excess phosphate-induced depletion of magnesium contributes to the enzyme, parathyroid, and bone changes.
Feinblatt et al. (1970) also observed that high phosphate/calcium ratios (in rats) cause similar lesions. They also demonstrated that phosphate infusions reduced the hypercalcemia caused by PTH administration, but did not alter its increase of urinary hydroxyproline secretion. Thus, their findings indicate that phosphate does not block bone resorption, but they assume that it increases bone mineralization. Comparable osteoporotic changes have been produced by excess dietary phosphate in adult intact and parathyroidectomized rats (G. Anderson and Draper, 1972) and in aging mice (Krishnarao and Draper, 1972) and rats (Draper et al., 1972). Lutwak (1974) has commented that such intakes are common in the American diet, and suggested that they might be contributory to the high frequency of osteoporosis and periodontal disease. In contrast, Berlyne et al. (1973b) have attributed the rarity of renal osteodystrophy in Israel to a low phosphorus intake.
11.3.2.2. Bone Mineralization
That phosphate loads might increase bone mineralization was first proposed by
F. Albright et al. (1932), who speculated that inorganic phosphate's antihypercalcemic effect (in hyperparathyroidism) was mediated by inhibition of bone resorption. Raisz and Niemann (1969) demonstrated this effect in vitro, and then showed that increasing the phosphate concentration of suspending media stimulated collagen synthesis by rat bone (Raisz, 1970). However, phosphate loading has stimulated PTH secretion, and failed to inhibit PTH-induced bone resorption, as indicated by continued excretion of bone minerals and hydroxyproline (Pechet et al., 1967; Feinblatt et al., 1970; Rasmussen et al., 1970). Despite its failure to suppress PTH-bone resorption, Pechet et al.(1967) reported that the neutral phosphate stimulated bone formation and mineralization. They explained this finding on the basis that considerable amounts of phosphate are bound by collagen and initiate crystal nucleation and growth (Glimcher and Krane, 1964).
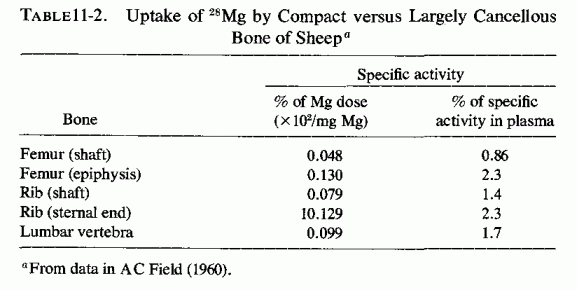
Availability of radioisotopes of magnesium (i.e., 28Mg) has permitted study of the influence of the metabolic activity of bone on its uptake and release of magnesium during short periods of time. Brandt et al. (1958) found considerable variability in skeletal 28Mg uptake by different bones of different dogs, and postulated that the rate of uptake is likely to be affected by many factors: growth, renal function, and the magnesium stores of the body. A. C. Field (1960) found that there was marked variation in magnesium uptake from bone to bone in sheep. It was greater in regions of rapid bone metabolism than in compact bone (Table 11-2). Using a radiographic procedure to measure the uptake of 28Mg in puppies, Glaser and Gibbs (1962) showed that the growing, actively metabolizing portion of bone (the epiphyseal line) concentrates most of the 28Mg that is taken up by bone, as compared with the diaphysis, the least active portion.
{kind=link}
In a study comparing predominantly the influence of age on the amount of magnesium mobilized from bone of magnesium-deficient rats (B. S. W. Smith and Field, 1963), there was relatively more magnesium lost per unit of mandible than per unit of femur. More magnesium was lost from the bones of the young rats (mandibular versus femoral magnesium loss = 33.3% versus 28.2%), but proportionally more was lost from the mandibles of the older rats (13.4% versus 9.5%). Parr (1957) confirmed the greater loss of magnesium from cancellous than from compact bone of magnesium-low calves. McAleese (1961) showed that epiphyses of magnesium lambs took up more 28Mg than did the diaphyses, indicating either more magnesium loss from the area of bone growth, its greater magnesium requirement, or both.
In a serial study of loss of magnesium from vertebrae, R. H. Smith (1959) amputated the terminal caudal vertebra at monthly intervals from magnesium-deficient and control calves, and found that the magnesium content of the bone ash dropped before the appearance of clinical signs of deficiency. Larvor et al. (1964a) showed that the diaphyses of magnesium-deficient calves lost less magnesium (compared with controls) than did the vertebrae. The ratio of vertebral magnesium in deficient versus control calves was 0.16:0.35; that of diaphyseal magnesium was 0.25:0.41. There was very little difference in bone calcium or phosphorus in the magnesium-deficient and control calves. B. S. W. Smith and Field (1963) found that magnesium-deficient rats lost relatively more magnesium from mandibular than from femoral bone. Minimal osteoblastic and alkaline phosphatase activity was found in alveolar bone of magnesium deficient rats (Trowbridge and Seltzer, 1967).
Aikawa (1965) demonstrated that the rate of bone uptake of 28Mg is influenced by the metabolic activity of the bone cells. Administration of insulin and glucose (Aikawa, 1960a) or of pyridoxine (Aikawa, 1960c) increased the bone uptake of 28Mg inhibitors of thyroid function of pyridoxine activity, or irradiation, decreased bone 28Mg uptake (Aikawa, 1960b; Aikawa and Reardon, 1963; Aikawa, 1965). MacManus and Heaton (1970) demonstrated that, in vitro, metabolically active bones release more magnesium to a magnesium-free medium than do bones whose enzymatic activity has been destroyed by aging. Heaton (1971) thus concludes that magnesium is released by a mechanism that is dependent on the metabolic activity of bone cells. (In the in vitro system, most of the magnesium released reflects establishment of a physicochemical equilibrium between the bone and its surrounding fluid.)
Bones with a high proportion of cancellous to compact bone (more metabolically active) develop clinically manifest osteopenia before predominantly compact (long) bones do. Thus, the greater loss of magnesium from such bones, relatively early in magnesium deficiency, might be clinically important, in view of magnesium's significance in so many enzyme systems (Reviews: Lehninger, 1950; Green and MacLennan, 1960; Heaton, 1976/1980). The intensification of magnesium deficiency by calcemic agents and phosphates, such as are commonly used in treatment of osteopenias and hypercalcemic states, might intensify bone matrix abnormalities and lead to the formation of hypermineralized bones with little matrix. It is possible such bones are similar to the brittle chalky bones and teeth seen in magnesium-deficient rats fed diets high in calcium, phosphate, and vitamin D Animals fed magnesium-deficient rations more similar to the human diet (high P/Mg and P/Ca) ratios tend to develop osteopenia. Rarely is the possibility that calcemic treatment of clinical osteopenias might intensify magnesium deficiency considered (Amiot et al., 1969; Durlach, 1971).
That skeletal magnesium is not readily mobilized in adult animals was suggested by the early work of Cunningham (1936a), who showed that bones from lactating cows with grass tetany and hypomagnesemia contained normal amounts of magnesium. Calves, however, kept on a diet low enough in magnesium (Mg/Ca=¼) to cause convulsions or death in 8 to 16 days, lost about two-thirds of their bone magnesium (Blaxter et al., 1954). Blaxter (1956) later evaluated the tissue magnesium changes in magnesium-deficient calves and found that soft tissue levels were not significantly depleted, but that there was a 56% loss of bone magnesium. His data suggested that the loss of skeletal magnesium takes place at the surface of the bone crystals, and that it occurs more readily in young than in old animals. Less severely depleted calves lost less bone magnesium, but more than did cows with lactation tetany (Parr, 1957), a condition associated with magnesium depletion.
In the case of rats, which continue to grow after they reach sexual maturity, the results are generally not as clear-cut. Breibart et al. (1960) found that young rats (20 to 30 days of age:44-100 g) exchanged 31-46% of their bone magnesium with 28Mg whereas 60 to 180 day-old rats (130-225 g) exchanged only 4-5% of bone magnesium. Young rats (90-110 g) that were kept on a magnesium-deficient diet high in calcium (Mg/Ca = 3.8/1500 mg/100 g diet) that maintained their growth, but at 1/6 the control rate, showed a pattern of distribution of injected 28Mg different from controls (Chutkow, 1965). Initially (within 3 minutes after the injection) there was prompt uptake of greater amounts of 28Mg (than in controls) by all tissues, including bone. Thereafter, most of the 28Mg was diverted to the soft tissues; the skeletal uptake of 28Mg did not exceed that achieved during the first few minutes. The study of A. C. Field and Smith (1964) was on the effect of magnesium deficiency on the uptake of 28Mg by mature rats (9-12 months old; avenging 400 g in weight), but cannot be directly compared with the Chutkow study (1965) because the Mg/Ca ratio was much lower: CaCO3: 75 parts, versus hydrated MgSO4 : 26 parts in controls, and absent from deficiency diets. They (Field and Smith) found that the bones of magnesium deficient rats took up less 28Mg than did the viscera (versus controls). The mandible took up relatively more magnesium than did the femur, the uptake of which was about equal to that of skeletal muscle. The relative specific activities (the ratio of that of the tissue to that of plasma, a measure of the proportions of exchangeable magnesium) of bone from the magnesium-deficient adult rats were less than in control rats, in contrast to the relative specific activities of the vital organs.
B. S. W. Smith and Field (1963) compared the amount of magnesium mobilized from the bones of 8-week-old male and female magnesium-deficient rats (180 and 140 g) with that from 9-to 12-month-old males (average weight: 400 g). They found that the young rats lost much more bone magnesium than did the old rats. The femurs of the magnesium-deficient young rats showed 28.2% magnesium depletion from femurs, as compared with controls; the mandibles showed somewhat more: 33.3% magnesium depletion versus controls. There was less loss of magnesium from the adult rats: 9.5% depletion in femurs; 13.4% depletion in mandibles versus controls. Martindale and Heaton (1964), however, found that mature rats, 4 to 5 months of age, lost bone magnesium rapidly during the first 15 days of deficiency, and then more slowly to reach about half the starting value after 62 days. The pattern of change was similar to that seen in blood plasma. These rats showed a significant rise in bone content of calcium and sodium, a finding in accord with the early studies (Orent et al., 1934), in which rats were given rations high in calcium. [Note that most magnesium-deficient rat diets are high in calcium, phosphate, and vitamin D (Review: Larvor and Durlach, 1971b).]
The hypocalcemia of severe magnesium depletion, which has been attributed to target organ unresponsiveness to PTH (or to failure of PTH release or secretion), has been explained by physicochemical factors involving ionic exchange of magnesium and calcium at the bone surface. Heaton (1971) has reviewed the evidence that bone magnesium is much more readily available than is bone calcium. (About a third occurs within the apatite crystals, the remainder being either adsorbed on the crystal surface or present in solution within the hydration shell around the crystals.) Duckworth and Godden (1941) showed that calcium exchanges for magnesium in the apatite crystal during magnesium depletion. Neuman and Neuman (1957) suggested that calcium ions can enter the extracellular fluid from bone only if the bone crystal takes up other cations (i.e., magnesium) to maintain electroneutrality. R. H. Smith (1961) speculated that the correlation of falls in plasma magnesium and calcium in magnesium-deficient calves might affect the availability of bone calcium. He observed that the fall in bone magnesium levels reflects that of serum magnesium, and that thus there is less extracellular magnesium available for exchange with calcium. Zimmet (1968) considered this possibility in interpreting the hypocalcemia of his magnesium-depleted patients, noting that Heaton and Fourman (1965) had suggested that magnesium deficiency interferes with release of calcium from bone. Larvor et al. (1964) showed that, during the early stage of magnesium deficiency in the calf, there is a slowing of the rate at which skeletal calcium exchanges with that in the blood. The postulate of Neuman and Neuman (1957) was proved when it was shown that addition of magnesium to an incubation medium increases the release of calcium from bone (Pak and Diller, 1969; MacManus and Heaton, 1970). The magnesium-induced release of calcium is accompanied by liberation of hydroxyproline (MacManus and Heaton, 1970), suggesting that magnesium is involved in bone turnover (Heaton, 1971).
In a 1950 review, Lehninger reported that virtually all phosphatases or phosphate-transferring enzymes are activated by magnesium. As early as 1931, Von Euler and Rydbom found that magnesium, fed to rats on a rachitic diet, increased their subnormal serum phosphatase levels. Snyder and Tweedy (1942) reported that severe experimental magnesium deficiency causes reduced serum alkaline phosphatase activity, an effect that has been verified in cattle and rodents (Larvor et al., 1964a; Heaton, 1965; Pimstone et al., 1966; Trowbridge and Seltzer, 1967; B. Smith and Nisbet, 1968; Hamuro, 1971; Elin et al., l971b; Loveless and Heaton, 1976). The observations that serum and skeletal alkaline phosphatase levels are low in acutely magnesium-deficient rats, and that addition of exogenous magnesium to the medium does not raise the enzyme level to that found in tissues of control rats, indicate that magnesium deficiency reduces the amount of phosphatase present, and not just its activity (Loveless and Heaton, 1976). Low bone levels of alkaline phosphatase have also been found in acutely magnesium-deficient rats by Trowbridge and Seltzer (1967) and Lai et al. (1975). Subacute magnesium deficiency in rats did not cause lowering of bone or serum alkaline phosphatase (Watchorn and McCance, 1937).
In a long-term magnesium depletion study (in patients who had undergone radical face and neck surgery for cancer), serum alkaline phosphatase levels gradually declined (to 1-2 Bodansky units) and did not increase with magnesium supplementation until the 56th day of repletion (Shils, 1969a). A shorter (1 month) study of healthy young men on a low-magnesium diet showed no reduction in serum alkaline phosphatase, even though their magnesium deficit was demonstrable by retention of large amounts of magnesium during repletion (Dunn and Walser, 1966). These volunteers did not develop hypomagnesemia; it seems likely that their bone stores of magnesium were sufficient to prevent interference with serum alkaline phosphatase activity. Possibly masking a (presumed) decrease in enzyme synthesis might be mobilization of alkaline phosphatase from the bone, to a lesser degree than that seen in neoplastic and bone diseases (Taswell and Jeffers, 1963; Moses and Spencer, 1963).
Low serum alkaline phosphatase activity was demonstrated in children with protein calorie malnutrition (R. Schwartz, 1956), a condition in which magnesium depletion has been identified. R. Schwartz (1956) has proposed that the very low serum alkaline phosphatase activity of such children can be correlated with decreased osteoblastic activity. Addition of magnesium to their serum increased the enzymatic activity, but not to the level found in normal children, an effect similar to that reported in studies of magnesium-deficient rats (Heaton 1965; Pimstone et al., 1966).
Low levels of serum alkaline phosphatase have also been found in adults with severe, long-term magnesium depletion (Hanna et al., 1960; Hanna, 1961b; Zimmet et al., 1968; Sutton, 1968; Muldowney et al., 1970; T. B. Connor et al., 1972), and have risen with magnesium infusions (Zimmet et al., 1968). They have also been reported in infants with hypercalcemia related to hypervitaminosis D and in other conditions associated with hypercalcemia (N. J. David et al., 1962). Since both excess vitamin D and calcium predispose to magnesium deficiency, the low alkaline phosphatase levels found in such patients might reflect a conditioned magnesium deficiency. Patients with bone involvement of neoplastic disease (who had hypecalcemia) had lower alkaline phosphatase levels than did those with normocalcemia (Moses and Spencer, 1963). In fact, the hypercalcemia preceded the lowering of enzyme levels (Griboff et al., 1954), possibly a reflection of calcium inhibition of phosphatase.
The genetic bone disorders associated with hypophosphatasia, and in which abnormal magnesium metabolism might play a role, are discussed elsewhere. One such disease, osteosclerosis, which is seen in infantile hypercalcemia [associated with hyperreactivity to vitamin D (Review: Seelig, 1969b) has been duplicated in pigs given 5 to 25 the antirachitic dose of vitamin D (Chinemene et al., 1976)]. On higher doses, the pigs developed hypophosphatasia. The few studies of magnesium in infants with the established syndrome have yielded conflicting results. However, one valuable study has been found that provides evidence suggestive of magnesium malabsorption in an infant with osteopetrosis, who had biochemical findings of hypophosphatemic rickets before high-dosage vitamin D therapy had been started, and whose alkaline phosphatase levels dropped from high to low during the eight months of vitamin D therapy (Pincus et al., 1947). A woman with magnesium-deficient latent tetany and rapidly progressive osteoporosis (Seelig et al., 1975), which was found due to renal magnesium wasting (Seelig et al., 1978), exhibited a sharp drop in her serum alkaline phosphatase following a period of supplementation with 25-OH-D3 during which her serum magnesium level fell further (unpublished data).
Another nutritional imbalance that has caused hypophosphatasia in several species, in association with hypercalcemia, is a normal calcium intake with three to four times as much phosphorus or more (Krook et al., 1975). This diet is considered one that causes nutritional secondary hyperparathyroidism and that is associated with progressive osteopenia. Not considered as a factor in this model is the magnesium deficit that is produced by phosphate loading. It is conceivable that the secondary hyperparathyroidism, the osteopenia, and the hypophosphatasia might all reflect magnesium depletion. Hamuro (1971) reported that on the first day of a high-phosphate, low-magnesium diet there was a slight increase in serum alkaline phosphatase levels in genetically diabetic mice. By days 4 to 6, the enzyme levels dropped to half the initial value. This decrease was not seen when the diet was supplemented with magnesium or when the phosphorus intake was reduced.
Pyrophosphatase, which also has an absolute and relatively high magnesium requirement (Magana et al., 1955; Kunitz and Robbins, 1966) was studied in erythrocytes of magnesium-deficient rats (Elin et al., 1971b). It took two weeks of a diet low in magnesium for red cell pyrophosphatase to drop and two weeks of repletion for it to return to control values. The serum alkaline phosphatase levels dropped more rapidly with magnesium deficiency and responded more quickly with repletion. The authors commented that the delay in pyrophosphatase response to magnesium deficiency and repletion is consistent with the slow fall in erythrocyte magnesium levels with its deficiency (Tufts and Greenberg, 1937) and the evidence that the amount of magnesium in the red cells reflects the magnesium status during their formation (Ginsberg et al., 1962). Heaton (1978) has surveyed the interrelations of magnesium with alkaline phosphatase, pyrophosphatase, and orthophosphatase activities. He has considered the controversy as to whether magnesium inhibits or activates pyrophosphatase activity and concluded that the experimental conditions influence the response of the enzymes to magnesium. The general view is now that magnesium stimulates the hydrolysis of pyrophosphate under normal conditions.
It is difficult to obtain precise data as to phosphatase levels, clinically, since the clinical chemistry laboratories report a single serum alkaline phosphatase figure, not distinguishing between that of skeletal and other (e.g., hepatic) origin. Several fractions have been differentiated (Keiding, 1959; Taswell and Jeffers, 1963). Where there is a disease that is likely to cause magnesium loss, and thus abnormal skeletal alkaline phosphatase activity, the high hepatic alkaline phosphatase values that derive from hepatic damage would obscure skeletal hypophosphatasia. Only research laboratories provide data on the differential alkaline phosphatase levels, and only rarely are pyrophosphatase levels obtained.
Robison (1923) postulated that bone alkaline phosphatase liberates inorganic phosphate from organic phosphates, with resultant localized increase in phosphate, which then precipitates the calcium. The in vitro studies that show that considerable amounts of phosphate are bound by collagen and initiate calcium crystallization (Glimcher and Krane, 1964) support the premise that interaction of phosphate with collagen plays a role in bone mineralization (Pechet et al., 1967). During bone growth and during osteolytic processes, the serum alkaline phosphatase activity increases (Griboff et al., 1954; Keiding, 1959). Possibly during new bone formation this reflects increased enzyme synthesis; during bone breakdown it might reflect increased enzyme release. On the other hand, both organic and inorganic polyphosphates inhibit calcium phosphate nucleation and precipitation (in collagen or bone matrix). Without an optimal amount of alkaline phosphatase to destroy the inhibitor, bone mineralization is impeded (Fleisch and Newman, 1961, Fleisch and Bisaz, 1962a,b). Subnormal synthesis or activation of enzymes that act to increase the mineralization process, by removing polyphosphate or pyrophosphate inhibitors, can be correlated with clinical conditions associated with abnormal bone formation and low phosphate levels. The most obvious condition is the uncommon genetic defect, hypophosphatasia, in which the magnesium status has not been explored, but that is characterized by unexplained convulsions in infancy not unlike those of hypomagnesemia, with and without hypocalcemia.
The abnormal high pyrophosphate levels found in serum and bone of infants and children with osteogenesis imperfecta, and the in vitro lowering of their bone biopsies' pyrophosphate content by addition of pyrophosphatase and magnesium suggest that skeletal hypopyrophosphatasia is likely to be an important factor in this disorder. The lowering of serum and urine pyrophosphates of such patients, with magnesium therapy, suggests that abnormal magnesium metabolism (possibly magnesium malabsorption or wasting) might be contributory.
Patients with bone disease, characterized by increased bone turnover (metastatic malignancy, hyperparathyroidism, hyperthyroidism, and Paget's disease) have all exhibited significantly increased urinary outputs of pyrophosphates, as well as of hydroxyproline. This increased pyrophosphate output might be an index of the amount of bone "metabolized" daily (Avioli et al., 1965). Considering this finding and the preliminary evidence that pyrophosphatase might be part of a control mechanism in both formation and resorption of bone, Tenenhouse and Rasmussen (1968) studied its activity in cell suspensions at a fixed physiologic magnesium concentration, at physiologic pH, and as influenced by PTH and CT. They found that PTH inhibits pyrophosphatase activity, and that CT reverses the inhibitory effect of PTH, effects that they considered to be mediated in part by altering the extracellular ionic environment. Orimo et al. (1970) demonstrated that CT administration to rats rapidly increases alkaline pyrophosphatase activity of bone, and suggested that it stimulates bone formation by removing the inhibiting pyrophosphate. These observations should be considered in light of the influence of magnesium on the secretion of both hormones, and on the response of target organs such as bone. It should be kept in mind here that the effects of magnesium deficiency on the hormones and bone depend on the duration and extent of the deficiency. Acute short-term magnesium deficiency increases PTH secretion. Long-term chronic deficiency decreases PTH release and bone response. High-dosage magnesium suppresses PTH secretion. The secretion of CT [which increases osteoblastic activity and decreases bone mineral mobilization (Review: S. P. Nielsen, 1974)] is stimulated by a low magnesium/calcium dietary ratio (Stachura and Pearse, 1970; Rojo-Ortega et al., 1971/1973) and by increased magnesium levels in vitro (Radde et al., 1968, 1970) and in vivo (Care et al., 1971; S. P. Nielsen 1971/1973; S. P. Nielsen and Jorgensen, 1972; Littledike and Arnaud, 1971).
Increased alkaline phosphatase activity has been demonstrated in the hyperplastic membrane of the thickened diaphysis and subperiosteal proliferation of magnesium-deficient rats (Bélanger et al., 1972), which also showed the more typical epiphyseal growth suppression. This observation supports the premise that the high level of the enzyme lowers that of the inhibiting polyphosphates, allowing for increased mineralization of the diaphysis. Why this magnesium-dependent enzyme should be found in such high concentrations in the membrane of the bone shaft of magnesium-deficient animals requires resolution. Similarly, more study is needed into why the increase in bone shaft alkaline phosphatase of magnesium deficiency should be associated with hyperplasia, resembling desmoid tumors, that was characterized by more fibrous tissue in parathyroidectomized animals, more bone formation when PTH was given, and less subperiosteal hyperplasia when estradiol (an alkaline phosphatase stimulator: Malinow et al., 1960) was given. Another puzzling observation is the association of osteogenic sarcomas with beryllium, which inactivates alkaline phosphatase, possibly replacing the activating magnesium (Grier et al., 1949; Aldridge, 1950).
The bits of evidence that patients with genetic bone dysplasia have abnormal (usually low) bone phosphatase levels, and that low magnesium levels lead to abnormal matrix formation and to defective osteocytic differentiation, suggest that normal magnesium utilization might be at fault. Evaluation of the magnesium status and bone phosphatase levels and activity of patients with genetic or neoplastic bone disease, and of the effect of magnesium on the enzyme activity of the biopsies, might prove worthwhile. If it would lead to prophylactic or therapeutic approaches remains to be seen.
Part III: Chapter 12
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
Bone-wasting diseases that are resistant to physiologic doses of vitamin D, calcium, or phosphate and that have been treated with pharmacologic doses of each or of combinations of mineralizing agents, are likely to be associated with magnesium deficiency. In some instances, initial magnesium inadequacy might be contributory to the osteopenia, as in hyperparathyroidism, secondary to malabsorption, with hemodialysis with low-magnesium water, or possibly in pregnancy. There is suggestive evidence that severe magnesium depletion (in utero), alone or with hypervitaminosis D, might participate in abnormal fetal bone formation that might find expression as fractures of low-birth-weight infants, osteogenesis imperfecta, or hypophosphatasia. In infancy and later in life, vitamin-D- or parathyroid-refractory osteomalacia or hypocalcemia might also have magnesium depletion as a contributory factor. Failure to correct the magnesium deficiency before use of calcemic therapy has failed to correct hypocalcemia. In those with osteopenia (to which magnesium deficiency has contributed), failure to correct that deficit before starting aggressive mineralizing therapy intensifies the imbalance.
Apart from the risk of thereby increasing the risk of extraskeletal damage and calcinosis, such treatment can adversely influence the skeletal system. It can lead to hypermineralization of bone or abnormal matrix, in some instances with exuberant osteoid formation. Marbleized or chalky brittle bones (such as have been described in rats on diets rich in calcium, phosphate, and vitamin D, and poor in magnesium) might develop. If the diet is high in phosphate but poor in calcium and magnesium, osteopenia has been seen. Low magnesium intakes, when severe, have been associated with desmoidlike tumors; whether the abnormal osteoid at sites of pseudofractures is a disorder of common etiology remains to be determined. It is provocative that severe magnesium depletion is most commonly recognized with vitamin-D- or PTH-refractory hypocalcemia, usually without bone wasting.
It has long been known that magnesium-deficient animals have arrested growth (Leroy, 1926; Kruseet al., 1932), but the precise nature of the bone abnormalities produced has not received much attention. In 1930, Huffman et al. reported that the ribs of magnesium-deficient calves are easily broken at the sternal ends, and that the specific gravity of the long bones is subnormal. Cunningham (1933) reported that rats on diets low in magnesium have narrowed epiphyseal plates, which contain few chondrocytes, and that the subepiphyseal region has few trabeculae. Duckworth et al. (1940) noted the fragility of the long bones of their magnesium-deficient rats and attributed it to abnormal matrix. Yamane and Singer (1953) observed that metaphyseal bony trabeculae are lost and that the zone of preliminary calcification just below the epiphysis is atrophic or absent in magnesium-deficient hamsters. Blaxter (1956), who found no histological evidence of abnormality of the calcification process in bones of acutely magnesium deficient calves, speculated that the matrix might be adversely affected. E. R. Morris and O'Dell (1961) postulated that magnesium deficiency interferes with cellular function of hard tissues, thereby preventing formation of normal matrix.
Since then, firm evidence has been obtained that magnesium deficiency does, in fact, interfere with normal formation of the matrix of both bones and teeth. Bernick and Hungerford (1965) found differences in staining characteristics that suggested that the ground substance of the magnesium-deficient matrix contains mucopolysaccharides that are less polymerized and less subject to normal calcification. They also compared the histologic differences between the epiphyses of the rats fed a magnesium-deficient diet for 19 days, and those of controls. They confirmed the early evidence that the cartilage of the epiphyseal plate of the heads of the tibias of the deficient rats is slightly narrower than normal. They found trabeculae, extending from the metaphysis into the diaphysis, that are shorter and wider than normal. The proximal epiphyseal cartilages of the deficient rats exhibit a slight decrease in number of proliferating cells, and relative increase in the number of hypertrophied cells, and a decrease in the width of the calcifying matrix. Clark and Bélanger (1967) also showed thinner epiphyseal plates in magnesium-deficient rats, and few chondrocytes. There were practically no new trabeculae at the subepiphyseal area, and the diaphysis contained immature matrix with small elongated osteocytes. This was confirmed in later studies (Hunt and Bélanger, 1972; Bogoroch and Bélanger, 1975).
Trowbridge and Seltzer (1967) investigated the effect of acute magnesium deficiency on the organic matrix of bone and dentin employing the uptake and tritiated proline to assess collagen formation,35SO4 to assess sulfation of protein polysaccharides, and measuring the biochemical localization of alkaline phosphatase in bones and dentin. They found minimal osteoblastic activity, with marked suppression of the amount of tritiated proline uptake in the collagen of the bone matrix in the deficient rats. Also, sulfation of glycosaminoglycuronoglycans was diminished in the osteogenic layer of the periodontal ligament, and reduced intensity of staining for alkaline phosphatase in the periodontal ligament and in osteoblasts suggests that magnesium deficiency decreases bone alkaline phosphatase; serum levels of the enzyme were also low. Hunt and Bélanger (1972) also showed that the cartilage matrix of magnesium-deficient rats appeared depleted in sulfated mucopolysaccharides. The underlying bone spicules of the epiphyseal plate of the tibia were thin and poorly ossified, as were the diaphyses.
Lai et al. (1975) have verified the reduction in bone magnesium, phosphatase, and matrix in magnesium-deficient rats and found hypermineralization with increased bone ash and calcium content. Their findings were more similar to those of Orent et al. (1934) and Watchorn and McCance (1937) than they were to those of Duckworth et al. (1940). The former two groups had fed their rats diets high in calcium and vitamin D; the latter used less calcium and did not indicate use of vitamin D. [Lai et al. (1975) did not specify the nature of the basic diet, but basic rat diets, provided currently from most firms, are rich in calcium, phosphorus, and vitamin D.] The bones were more brittle than those of control rats.
Bone matrix implanted in skeletal muscle of magnesium-deficient rats, and controls fed the same commercially supplied deficient diets, but supplemented with 265 mg/100 g of diet showed marked differences in bone growth and development (Belanger and Robichon, 1975). There was osteoporosis in the lumbar vertebrae of the magnesium-deficient rats, and very little bone formed on either the inside or the outside of the implants. Only 2 of the 15 rats that survived 3 weeks formed new trabeculae, and these were fibrillar and poorly mineralized. At several sites, there were small amounts of bone tissue mixed with islands of chondrocytes, surrounded by precartilaginous or poorly differentiated matrix. Further outward from the implant there was an "envelope" of fibroblastlike cells and collagen that separated the implant from the muscle. In half of the specimens, there was invasion of the implant by thin cartilage wedges, coming mostly from the periphery of the implant. Near these wedges, there was deterioration of the implant matrix. In contrast, the magnesium-supplemented rats had formed well-mineralized trabecular bone inside and outside the implant. In a few areas, cartilage or a mixture of cartilage and bone had formed.
Paradoxically, magnesium-deficient animals have exhibited excess (abnormal) bone growth in addition to osteopenia. Generalized medullary bone growth (osteomyosclerosis and periosteal tumors of the desmoid type occurred at the femoral linea asperea in severely magnesium-deficient rats (Bélanger and Hunt, 1971/1973; Hunt and Bélanger, 1972). (This is a prominent longitudinal ridge on the middle third of the bone, with rich vascular supply, and concomitant sites of accretion and osteocytic osteolysis, which indicate that it is an area of considerable metabolic activity.) The degree of magnesium deficiency was critical in the formation of both the periosteal tumors and intramedullary bone; both abnormalities were produced only in severely magnesium-deficient rats, but irrespective of the concentration of bone magnesium. The investigators speculated that matrix and bone cells could be differentially depleted of magnesium, and that the bony overgrowth was related to changes in the magnesium concentration in the organic phase of bone. The periosteal tumors rapidly disappeared when the rats were supplemented with magnesium. This was interpreted as indicative of magnesium depletion-induced accumulation of cells unable to differentiate properly, possibly as a result of enzymic malfunction. Deficient rats that developed fibrous hyperplasia showed high concentration of alkaline phosphatase activity throughout the hyperplastic membrane (Bélanger et al., 1972a).
Although the periosteal desmoid tumor was first shown to be a characteristic of magnesium deficiency by this group (Hunt and Bélanger, 1972), the authors noted that Duckworth et al. (1940) had referred to "disordered growth of the organic matrix of leg bones" in their magnesium-deficient rats that might have been a comparable phenomenon. Lai et al. (1975) later observed that 10 of their 11 magnesium- deficient rats had tumorlike femoral exostoses.
These tumorlike growths resemble that described by McCance (1946) in an adolescent girl who developed weakness and hypophosphatemic vitamin-D-resistant osteomalacia at the age of 15. She had multiple spontaneous pseudofractures and callus formation of her long bones (Looser's nodes), and had a tumor on the shaft of the tumor. Histologic examination showed abnormal osteoid tissue that was not considered neoplastic. Metabolic balance studies, done while the patient was receiving about 2000 units of vitamin D daily, showed substantially negative balances of calcium, phosphorus, and magnesium. On a daily magnesium intake of about 230 mg, she lost an average of 25 mg/day over a 7-day period. Massive vitamin D therapy (500,000 units/day) greatly improved her retention of calcium and phosphorus but improved the magnesium retention only slightly. The vitamin D was stopped when signs of toxicity developed (after a month), and her magnesium retention improved markedly (to + 40 mg/day). When her magnesium intake was increased (to 390 mg/day) she went into strongly positive magnesium balance (+ 90 mg/day) and showed steady clinical improvement.
It is provocative that similar exostoses, described as irregular subperiosteal new bone formation or exuberant callus, have been reported in patients with hypophosphatasia (Schlesinger et al., 1955; Currarino et al., 1957), a condition postulated to be related to magnesium depletion. Hypophosphatemic rickets has also been associated with profound weakness and Looser's nodes in an adult, who also had a lengthened QT interval (Milne et al., 1952). The authors attributed the weakness and abnormal ECG to her hyperkalemia. The bone and cardiac manifestations might also have had magnesium deficiency as a common cause.
Hunt and Bélanger (1972) found that parathyroid activity influenced the nature of the bone tumor produced by experimental magnesium deficiency. Parathyroidectomized magnesium-deficient rats had a large tumoral mass that consisted of layers of fibrous tissue on the outside, then cartilage, and an internal layer of bone. Administration of PTH to these animals reduced the amount of cartilage, which then appeared as small peripheral isolated units, and resulted in abundant growth of medullary bone throughout the central cavity of the femur and tibia. In view of the estrogen/PTH antagonism in bone accretion and resorption (Ranny, 1959; Review: Seelig and Lehr, 1971/1973), the observation that ovariectomy, with and without estradiol administration, modified the incidence and severity of skeletal lesions caused by magnesium deficiency (Bogoroch and Bélanger, 1975) is provocative. Although the reduction in diaphyseal width of magnesium-deficient ovariectomized rats did not differ significantly from that seen in intact deficient rats, some of the ovariectomized rats showed greater subperiosteal hyperplasia. Estradiol-treated magnesium-deficient rats had a quarter the incidence of subperiosteal hyperplasia seen in untreated magnesium-deficient rats. Possibly related is the fourfold increase in plasma alkaline phosphatase activity produced by estradiol treatment of chickens (Malinow et al., 1960), since alkaline phosphatase synthesis and activity are magnesium dependent and the enzyme is involved in bone metabolism.
Bone tumors have also been experimentally produced by beryllium (Janes and McCall, 1975), which inhibits alkaline phosphatase, an inhibition that is partially reversed by adding magnesium (Aldridge, 1950; Grier et al., 1949). It is thus of interest that both in human and experimental osteogenic sarcoma, magnesium bone tumor levels were low (Janes et al., 1972; Jones and McCall, 1975). This brings us to the discussion by Hunt and Bélanger (1972) of the significance of their observation of the osteomyelosclerosis and subperiosteal tumors of their magnesium-deficient rats. They noted that the deficiency-induced lesion seemed to correspond to the periosteal desmoid described in the Catalogue of Tumors of Bone and Cartilage (Spjut et al., 1969), which is described as midway "between a true tumor and nontumorous connective tissue hyperplasia." They also noted that the occurrence of osteomyelosclerosis occurs in human disease, frequently in association with certain forms of leukemia and other blood dyscrasias and that leukemia has occurred in magnesium-deficient rats (McCreary et al., 1967; Battifora et al., 1968).
Thus, magnesium deficiency causes major metabolic disturbances of the bone that can lead to osteopenic yet hypermineralized brittle bones as well as hyperplasia and might even participate in an early neoplastic process. The degree of the deficiency as well as the concomitant dietary imbalance and hormonal responses, affects the nature of the lesion produced. Possibly much of its direct effect on bone is mediated by its effect on synthesis or activation of phosphatases, by its effects on bone matrix, and by its influence on differentiation of the bone cells.
The failure of maintenance of fetal magnesium levels at the expense of the mother can result in responses in PTH and CT secretion that can influence fetal bone growth and development. Both the parathyroids and the C cells are functional early in fetal life. They are influenced by the maternal magnesium and calcium levels, both of which have been shown to have a tendency to below. Both cations, unlike PTH and CT, can cross the placental barrier, and thus must be controlled by fetal parathyroid and C-cell responses. This homeostatic control is mediated by their effects on fetal bone. Fetal rat bone, kept in a medium low in magnesium (0.3 mM), showed less release of tagged calcium when exposed to PTH (Raisz and Niemann, 1969), an effect like that seen in the intact magnesium-deficient experimental animal or human. Fetal bones in media high in magnesium (4.3 mM) showed the same release of calcium from bone as did bones kept in physiologic magnesium and calcium concentrations when PTH was added. Bone resorptive activity of human fetal parathyroids has been demonstrated as early as 12 weeks gestation, at which time there are secretory granules (Scothorne, 1964). Differentiation of cellular organelles are manifest later in fetal life (Altenahr and Wohler, 1971). That fetal thyroid tissue can secrete CT early in gestation is suggested by the better growth of fetal chick bones in the presence of 8-day fetal chick thyroid tissue than in its absence (Gaillard and Thesingh, 1968). There is increased fetal CT secretion in rats toward the end of gestation (Garel, 1970; Feinblatt and Raisz, 1971). Calcium infusion in the fetus has suppressed the hypocalcemic effect of CT (Garel, 1970). It has been suggested that this effect might be mediated by inhibition of bone resorption, as in the adult. Exposure of fetal rat thyroid tissue in vitro to increasing concentrations of calcium (to 2 1/2 times normal levels) increased the release of CT; it inhibited PTH stimulated calcium release from fetal bone threefold (Feinblatt and Raisz, 1971). When both calcium and magnesium concentrations were physiologic (Ca/Mg = 1.0/0.8 mM) additional CT caused some inhibition of PTH-stimulated release of 45Ca from fetal bone. When the magnesium concentration was reduced to 0.4 mM or raised to 1.6 mM a slight increase in CT secretion resulted, which increased the percentage inhibition of release of calcium slightly. When magnesium was further raised to 3.2 mM the CT-induced percentage inhibition of calcium release rose a little more, although none of the changes were sufficient to be considered significant. There has been considerable experimental evidence that CT increases bone calcification and new bone formation directly (Matthews et al., 1972; Wase et al., 1967; Pallasch, 1968; Ziegler and Delling, 1969; Delling et al., 1970; Gaillard and Thesingh, 1968; Salomon et al., 1973), independent of its counteraction of demineralization. Thus, the lowering of plasma levels of calcium, magnesium, and phosphorus in response to CT (Garel et al., 1968, 1969; Garel and Barlet, 1974) might indicate utilization of those elements in bone formation. The high CT levels in the fetus are likely to take part in bone growth and calcification (Samaan et al., 1973b).
Magnesium deficiency has been implicated in placental and fetal abnormalities. Placental calcification has been reported in magnesium-deficient rats (Dancis et al., 1971). Since most rat diets are rich in calcium, phosphorus, and vitamin D, further work is necessary to determine how much of the placental damage in that study might have been caused by the other nutrients. Hypervitaminosis D during pregnancy has been implicated in human and experimental placental damage. In rats, it has also caused fetal bone damage (Ornoy et al., 1969). That gestational hypervitaminosis D (which increases magnesium loss) causes both placental and fetal bone damage is provocative, but does not separate the possible direct effect of vitamin D toxicity from the presumed effect of magnesium deficiency on the placenta. That magnesium deficiency causes bone damage has been clearly demonstrated after birth. Vitamin D excess, given to pregnant rabbits, has caused premature closure of the fontanels, osteosclerosis, and palatal abnormalities (Friedman and Mills, 1969). Nonetheless, the levels of the 25-OH-D3 metabolite of young of rabbits given toxic doses of vitamin D have been subnormal (Mehlhorn et al., 1977), suggesting abnormality in vitamin D metabolism under these conditions.
Detailed study of the fetal bone abnormalities caused by toxic dosage of vitamin D in pregnant rats (40,000 units D2) showed that the fetuses had 61% decreased bone ash by the 21st day of gestation, shortened thin diaphyses, and abnormal epiphyseal cartilage. Pups of rats given half as much vitamin D (Ornoy et al., 1972) had bone deformities consisting of kyphoscoliosis and distortions of the long bones. There was less osteoid in the metaphyses, there were many metaphyseal fractures, and diaphyseal bone was short, distorted, and with much thinner than normal periosteal bone. By the 30th postnatal day, some of the pups had epiphyseal fractures. The authors observed that the prenatal vitamin D excess resulted in lasting defects in bone formation and imperfect healing of fractures in the newborn, that resembles some of the characteristics of osteogenesis imperfecta. When prenatal vitamin D excess causes osteopenia, it is possible that magnesium deficiency might complicate the picture, in that it might militate against CT release, high intakes of magnesium (like hypercalcemia) stimulating CT secretion. It should be kept in mind that fetal magnesium stores are likely to be suboptimal in magnesium-deficient mothers. Experimental magnesium deficiency has caused bone lesions (supra vide). Epiphyseal separation and osteochondrosis have been reported in premature infants (Griscom et al., 1971) and have been reported in older children with magnesium deficiency (Miller, 1944; Klingberg, 1970) or with the bone lesions of celiac disease of children (Parsons, 1927) or adults (Bronsky, 1970; Prost et al., 1972), which has been associated with either magnesium or vitamin D deficiency or both (Prost et al., 1972). It has also been associated with "pseudohypoparathyroidism" with parathyroid hyperplasia and hyper- rather than hypoparathyroidism, and is resistant to the action of PTH, further suggesting magnesium depletion.
Low-birth-weight infants, who might reflect intrauterine malnutrition rather than prematurity, are especially prone to magnesium depletion. The pathogenesis of the lesions described in this section is difficult to understand and interpret, conflicting findings having been reported and many factors interrelating. For example, magnesium depletion can cause decreased release of PTH and decreased target organ responsiveness (Fig. 12-1.). Yet acute magnesium deficiency has increased PTH release and chronic suboptimal magnesium intake has caused parathyroid hyperplasia (Larvor et al., 1964a). Vitamin D excess causes hypercalcemia, which can increase CT secretion and cause osteosclerosis(Fig. 12-2) or can cause magnesium loss and osteopenia (Fig. 12-1). Magnesium deficiency in infancy can cause hypocalcemic tetany and can be involved in vitamin D refractoriness. Both magnesium and vitamin D deficiency can cause fetal, neonatal, and later osteopenias. Yet mothers with presumptive magnesium deficiency and placental pathology have given birth to infants who developed osteosclerosis almost indistinguishable from that of rodents with hypervitaminosis D. Conversely, mothers with intestinal malabsorption, which probably interfered with absorption of both magnesium and vitamin D, gave birth to infants with congenital rickets. Furthermore, low-birth-weight infants have been shown to require vitamin D supplementation, above that in their fortified formula to avoid rickets (Lewin et al., 1971). It should be recalled, here, that magnesium deficiency increases vitamin D requirements. Vitamin D supplements have increased, significantly, plasma 25-OH-D3 levels in maternal, cord, and neonatal blood (Belton et al., 1975, 1977). The possibility of abnormal vitamin D utilization with gestational excess (Mehlhorn et al., 1977) should be kept in mind, low levels having been seen.
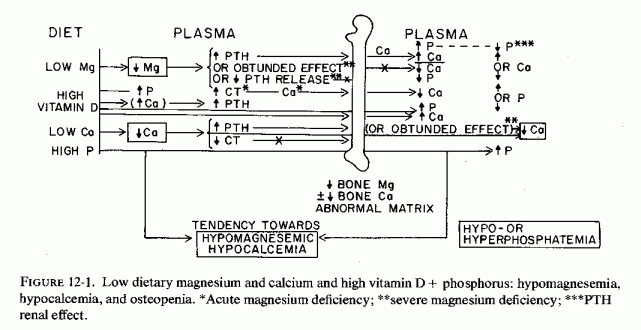{kind=link}
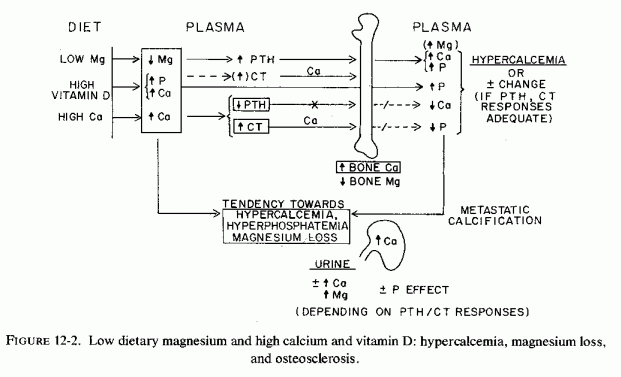{kind=link}
Experimental evidence has been presented that suboptimal supply of magnesium to the fetus can stimulate fetal PTH secretion. Whether it can contribute to abnormal vitamin D metabolism in the mother or the fetus should be explored. The degree to which maternal and fetal bone stores are utilized (under the influence of PTH secretion and vitamin D administration) probably depends upon the ratio of PTH and calcitonin (CT) levels and the metabolism of vitamin D, which are affected by both calcium and magnesium levels. Although fetal osteosclerosis has been correlated with excessive vitamin D, calcium, and phosphate administration, magnesium-deficient fetuses are also at risk of bone loss or defective formation.
Indirect evidence that this can be so derives from the osteoporosis, epiphyseal separation, poorly mineralized subperiosteal new bone, enlargement of costochondral junctions, metaphyseal cupping, and spontaneous fractures in three premature infants of women with placental pathology. Two of the mothers were preeclamptic and one was a young multipara who had two previous premature deliveries (Griscom et al., 1971). All of the women were probably magnesium deficient, two as a result of bearing twins and having preeclampsia, and one because of frequent pregnancies at a young age. That the infants might also have been magnesium deficient is suggested by the fact that two were survivors of twin pregnancies and one was premature. The twins who did not survive had been stillborn in one instance and had died at 10 hours in the other, the latter with thymic involution (such as is seen in infants with long-standing intrauterine distress) and with microfocal myocardial necrosis. The bone disease of these three infants was diagnosed a week before death: a few days after sudden cardiac arrest at 71 days in one, and two months after a cardiac murmur was diagnosed at one month in another. Additional suggestive evidence that magnesium deficiency might have been present was the severe anemia that developed in all three, such as has been produced in the young by experimental magnesium deficiency in pregnant rats (Cohlan et al., 1970) and in nonpregnant rats (Elin et al., 1971b; Elin, 1973, 1976/1980)
Griscom et al. (1971) pointed out that demineralization of bone may not be rare in low-birth-weight infants in the early weeks or months of life. The first such case, an atrophic newborn infant with osteogenesis imperfecta in association with arterio sclerosis, was reported by Johansson (1921-1922). Dystrophic osteomalacia of prematurity has been reported from France (Boissiere et al., 1964), and is characterized by icterus and pneumonitis as well as by bone disease. Griscom et al. (1971) found many similarities in the three infants they reported to those of the French infants (Boissiere et al., 1964). The disorder usually does not become manifest until the third month of life and commonly appears in twins. Fractures, subperiosteal new bone, and osteoporosis characterize the disease. However, only one of the three American infants of Griscom et al. had icterus, and that to only a slight degree. It was seen in 22 of 26 of the French infants. The American infants also did not present with hypocalcemic tetany, such as was reported from France. Griscom et al. (1971) considered the picture to reflect a metabolic, probably nutritional disorder other than rickets, and considered it likely to be fairly common among premature infants.
Another premature infant that developed rarefaction of ribs and scapula and spontaneous rib fractures by the third month of life, and also had anemia considered typical of prematurity, was diagnosed as rachitic (Keipert, 1970). This infant was the fourth child born in a difficult labor to an apparently normal mother. Intermittent apneic attacks began at 11 days. Despite vitamin D supplementation of 1,400-800 IU/d, some evidence of rickets persisted at nine months of age. The author commented that fractures are more common in rachitic than in normal bones, but observed that nonrachitic premature bones are also easily traumatized. He noted that the subperiosteal proliferation of prematurity is not related to vitamin D deficiency , and that Eek et al. (1975) found such changes earlier in premature infants fed cows' milk than in breast-fed prematures. Eek et al. (1957) postulated that double periosteal contours appeared in such infants when deposition of minerals increased after a period of poor mineralization. Tsang et al. (1977) have reviewed data on the abnormal and delayed skeletal mineralization in very low-birth-weight infants. Their group has shown that extrauterine bone mineralization lags significantly in such infants (Minten et al., 1976; Steichen et al., 1976).
It is provocative that the low-birth-weight infants who develop bone lesion rarely exhibit symptomatic hypocalcemia, such as is seen in those free of osteopenia. Possibly fetal hyperparathyroidism had mobilized fetal bone calcium. However, an alternative possibility must also be considered, that of the response of maternal, fetal, and neonatal C cells to changes in calcium and magnesium levels. High CT levels might contribute to both low plasma levels of calcium and magnesium, increasing bone mineralization.
The first clue to the vitamin-D-sparing effect of magnesium administration was provided by Huffmanet al. (1930), who found that a rachitic calf recovered when magnesium carbonate was added to his diet, which was low in calcium but adequate in phosphorus. When it was withdrawn, the abnormal signs recurred. His ribs were soft and broke easily. Calves fed whole milk for 45 days, after which they were given only skim milk and grain [a rachitogenic ration, with a high phosphorus/calcium ratio (Huffman et al., 1930, 1935)], developed hypocalcemia and hyperphosphatemia by 95 days. Magnesium carbonate administration, alone, did not cure the rickets, but when it was given with suboptimal amounts of vitamin D, the biochemical and clinical signs of rickets were corrected. The ash and mineral content of the bones indicated better utilization of calcium and phosphorus when magnesium supplements were given. R. H. Smith (1957, 1961) observed that magnesium supplementation of milk-fed calves that had developed hypocalcemia restored normal serum calcium levels even without vitamin D supplementation and speculated that magnesium might exert its effect on bone/extracellular equilibrium (of calcium). Magnesium-deficient calves had hypocalcemia requiring 70,000 IU/day to correct (R. H. Smith, 1958).
Baby pigs developed rickets on a normally balanced calcium and phosphorus intake (0.8% and 0.6% of the diet, respectively, when they were given less than 100 IU of vitamin D/kg of diet (E. R. Milleret al., 1964b). Most exhibited a moderate fall in plasma magnesium, even on what was shown to be optimal magnesium intakes: 350 ppm of diet (E. R. Miller et al., l965a), particularly in those that developed tetany. All developed hypophosphatemia and hypocalcemia. Balance studies showed that the vitamin-D-deficient pigs absorbed magnesium, calcium, and phosphorus poorly (E. R. Miller et al., 1965b). Doubling the magnesium intake of pigs given no vitamin D improved their weight gain, and prevented the mortality (that resulted in deaths of three of the four vitamin-D-deficient pigs on the standard magnesium intake) but neither prevented their rickets nor corrected their hypocalcemia or hypophosphatemia (E. R. Miller et al., 1964b). Thus, like most human infants, baby pigs require exogenous vitamin D to prevent rickets. Increasing the magnesium intake exerts a partially sparing effect on vitamin D requirements but cannot replace it.
On the other hand, when vitamin D supplements 18-fold higher than the antirachitic amount are given to baby pigs, the greatest strength and elasticity of the femur is obtained with an optimal magnesium intake of 325 ppm (E. R. Miller et al., 1965b). Analysis of rib ash showed no significant effect of low dietary magnesium on percentage of calcium or phosphorus, but a significant reduction in percentage of magnesium. Pigs on low-magnesium intakes, however, exhibited significantly less breaking strength and elasticity (Fig. 12-1). Since the elasticity of the bone is a function of the amount of matrix, the drop in elasticity-but not in bone ash, calcium, and phosphorus of the magnesium-deficient vitamin-D-loaded pigs-reflects the drop in bone magnesium, which is necessary for normal bone matrix formation.
Rats, which are the most commonly employed laboratory animals and with whom most vitamin D and magnesium interrelationships have been studied, differ markedly from the ruminants, pigs, and people as regards their susceptibility to rickets and their response to magnesium deficiency. They do not develop rickets, even when given no vitamin D supplementation, unless they are given diets high in calcium and low in phosphorus (Steenbock diet, cited by Shelling and Asher, 1932). McHargue and Roy (1930) showed that rats fed a normal diet, not supplemented with vitamin D and not exposed to ultraviolet light, remain free of rickets. Au and Raisz (1965) confirmed that rats not supplemented with vitamin D do not develop rickets unless their diets have a high ratio of calcium to phosphorus (0.8% Ca/0.1% P). Rats on high phosphorus to calcium ratios (0.1% P/0.03% Ca) had decreased bone density, but not rickets. The effect of high (14,500 ppm) and normal (6,500 ppm) intakes of calcium fed to groups of rats fed a normal amount of phosphorus (6,100 ppm), but low in magnesium (30 ppm), was studied by Rayssiguier and Larvor (1974a). By the tenth day of the low magnesium intake, all of the rats had hypomagnesemia and low levels of magnesium in their bones. Those that had been given high calcium diets the longest (17, versus 10 days), had the lowest bone magnesium levels. Since high calcium intakes interfere with intestinal absorption of magnesium and increase its urinary output (Reviews: Heaton, 1971; Larvor and Durlach, 1971b; Seelig, 1971), the rats made susceptible to rickets might have been magnesium deficient. Furthermore, vitamin D is necessary for optimal intestinal absorption of magnesium in rats (Meintzer and Steenbock, 1955), as well as in pigs (supra vide) and other species (Schachter and Rosen, 1959; Worker and Migicovsky, 1961) including man. Despite the defective magnesium absorption of vitamin D deficiency, the early rat studies showed high magnesium/calcium ratios in rachitic bones (possibly a reflection of the high osteoid/mineral ratio of such bones). McHargue and Roy (1930), who cited the early studies (Malcolm, 1904; Mellanby, 1926), found that exposure of rats to ultraviolet light for only three to five minutes daily or every other day resulted in better weight gain (than of nonirradiated litter-mates), but in significantly lower bone and total carcass magnesium levels. This work was done before it was realized that magnesium is an essential mineral, and the authors speculated that the beneficial effects of ultraviolet ratio might be the result of eliminating excess magnesium.
Supplementation with vitamin D of rats made rachitic by low phosphorus corrects the hypophosphatemia and heals the rickets (Tanaka and DeLuca, 1974), an effect attributed to a vitamin-D-dependent phosphate transport mechanism (DeLuca, 1976). In 1941, Harrison and Harrison showed that vitamin D increases renal tubular reabsorption of phosphate. Possibly vitamin D's increase of the intestinal absorption of magnesium might also play a role, magnesium deficiency having been shown to exert a phosphaturic effect, even in parathyroidectomized rats (Ginn and Shanbour, 1967). VonEuler and Rydbom (1931) noted the antirachitic effect of adding magnesium to a rachitic diet fed to rats and considered this effect possibly due to magnesium-induced increase in serum phosphatase activity.
In contrast, magnesium deficiency decreases responsiveness to vitamin D in ruminants and rats (R. H. Smith, 1961; Larvor et al., 1964b; Lifshitz et al., 1967a,b). Magnesium-deficient calves required 70,000 IU/day of vitamin D to attain normocalcemia; physiologic doses of vitamin D were not effective (R. H. Smith, 1958). Similarly, Lifshitz et al., (1967b) found that magnesium-deficient rats did not develop a calcemic response to 100 IU of vitamin D a week, but did when the vitamin D dosage was increased 10-fold. Their studies suggested that the poor response of serum calcium in magnesium-deficient rats, to physiologic doses of vitamin D, was due to decreased mobilization of calcium from the skeleton.
Whether impaired mineral mobilization in association with high calcium and vitamin D intake might account for the osteosclerosis seen in rats given intermittent high doses of vitamin D (Storey, 1960), and is mediated by magnesium-deficiency- induced abnormal bone development, deserves study. Storey (1960) observed that large daily doses of vitamin D inhibited endochondrial growth in rats, caused bone resorption and, later uncalcified matrix (osteoid), such as is seen in rickets. When the vitamin D was given intermittently, dense metaphyseal bone was formed in striations, which contained abnormal cartilage, changes resembling those seen in osteopetrosis. It is noteworthy, thus, that comparable changes were seen in infants and children with infantile hypercalcemia, commonly with the supravalvular aortic stenosis syndrome of clinical vitamin D overdosage. Since vitamin D excess causes magnesium loss, it is not surprising that its use (i.e., in milk, which also delivers ample calcium and phosphate) can produce changes in the matrix, such as is seen in magnesium deficiency as well as bone hypermineralization.
Lifshitz et al., (1967b) noted the lag between the time a physiologic dose of vitamin D was given and the calcemic response, and suggested that magnesium's mediating effect might be in its transformation to another form. Since then, it has been demonstrated that vitamin D is hydroxylated to active steroid hormones (e.g., in liver and kidney), and that some of the enzymatic steps require magnesium (Norman, 1968, 1971; DeLuca, 1969; Horsting and DeLuca, 1969; Norman et al., 1975/ 1977). Its deficiency in rats has interfered with the activity of the 1, 25-(OH)2D3 on calcium mobilization from bone, but has not prevented its enhancement of intestinal calcium absorption (Rayssiguier et al., 1974b, 1975).
The cited experimental evidence that magnesium deficiency causes relative refractoriness to vitamin D, very high doses being required for a calcemic response, and that magnesium repletion restores the responsiveness to physiologic doses (supra vide), is reflected by the refractoriness of hypomagnesemic patients to vitamin D. It suggests that the occasional report of correction of vitamin-D-refractory rickets by magnesium might be indicative of the need to evaluate all patients with vitamin-D-unresponsive bone disease for their magnesium status. Conversely, Durlach (1969, 1971) pointed out that patients with latent tetany of magnesium deficiency require less vitamin D when their magnesium deficit is repaired and must have their serum calcium monitored to avoid damage caused by hypercalcemia.
The magnesium/vitamin D/calcium/phosphorus interrelationships are particularly complex. Focusing only on magnesium is unrealistic. Disregarding magnesium is equally unrealistic. Considering only the magnesium/vitamin D interrelationships, if there is a deficiency of magnesium in infancy, for example, there is likely to be impaired response to vitamin D (and to PTH) with resultant hypocalcemia. However, we cannot ignore the hyperphosphatemia of infancy, which is contributed to by the cows' milk and the hypoparathyroidism, and which is enhanced by vitamin D therapy of the hypocalcemia. Thus, in attempting to correct infantile hypocalcemia by calcium loads and calcemic agents a vicious cycle can be established that causes direct loss of magnesium and might damage the area of the renal tubules where magnesium is actively reabsorbed (Fig. 12-4).
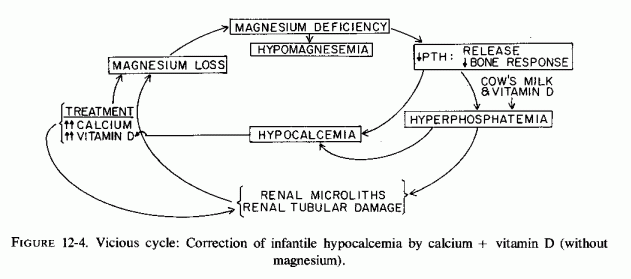{kind=link}
Hypophosphatemic (vitamin-D-dependent or vitamin-D-refractory) rickets is now the most common cause of rickets in children, and the pathogenesis is still obscure (Cohanim et al., 1972; Brickman et al., 1973). The syndrome was first attributed to hyperparathyroidism secondary to malabsorption of calcium (Albright et al., 1937), and then to an often familial intrinsic renal tubular defect of phosphate reabsorption (B. Robertson et al., 1942; Dent, 1962; Fanconi, 1955; Frame and Smith, 1958; Barbouret al., 1966). Despite the hypophosphatemia, biopsy of the epiphyseal area showed normal content of phosphate (Kuhlman and Stamp, 1964) and also above normal levels of bone alkaline phosphatase, hypertrophic cartilage cells, and thick areas of uncalcified osteoid. Subsequent work has confirmed both hyperparathyroidism (Lafferty et al., 1962; Riggs et al., 1969), usually secondary to intestinal malabsorption (Blackard et al., 1962; Falls et al., 1968; Reitz and Weinstein, 1973), and a genetic X-linked defect in renal tubular reabsorption of phosphate (Glorieux and Scriver, 1972; Glorieux et al., 1973; Scriver, 1973). T. F. Williams (1968) commented on the apparently simple genetics but multiorgan sites of expression in familial hypophosphatemic vitamin-D-resistant rickets. He called for a unifying way to explain the: (1) decreased renal tubular reabsorption of phosphate, (2) decreased intestinal reabsorption of calcium, (3) bony abnormalities, including both osteomalacia and overgrowth, and (4) improvement of calcium absorption and rickets, but not the phosphaturia, with large doses of vitamin D.
Possibly a form of the genetic defect, isolated magnesium malabsorption, is contributory, and might even be a common denominator. This is a point requiring intensive study, and not by measurement of serum magnesium levels. Analysis of bone biopsies for phosphatase and magnesium levels, and metabolic balance studies to ascertain the percentage absorption of orally administered magnesium might be useful. Since such patients are commonly loaded with calcemic agents and phosphates in the attempt to correct their hypocalcemia and hypophosphatemia, and such treatment has increased renal calcinosis, determination of percentage renal retention of magnesium might not be a good index of magnesium depletion. Renal magnesium wasting might result from formation of renal tubular microliths, with damage to the ascending limb of the loop of Henle, where active tubular reabsorption of magnesium takes place. This would perpetuate a magnesium deficit caused by intestinal malabsorption of magnesium.
Magnesium deficiency might be involved in several facets of vitamin D resistance. Both hyperparathyroidism and hypomagnesemia have been implicated in hypophosphatemia (Review: Knochel, 1977), and since familial hypophosphatemia has been found in vitamin-D-resistant rickets in infants and adults (Stickler, 1969; Arnaud et al., 1970; Morgan a al., 1974), there might be a common denominator. There have been several studies that demonstrate abnormal magnesium metabolism and levels, and a few that have shown clinical and biochemical improvement with magnesium therapy.
McCance (1946) reported negative magnesium, calcium, and phosphorus balance in a girl whose vitamin D resistance, osteomalacia, and pseudofractures developed during adolescence. Rosen and Finberg (1972, 1973) found strongly negative magnesium balances in children with active vitamin-D-dependent rickets, which became strongly positive when they had been healed as a result of administration of 25(OH)D3 an active vitamin D metabolite. However, despite negative magnesium balances during the active phase of the disease, serum magnesium levels were within normal limits. Among the conditions found to be associated with low total and ultrafiltrable magnesium levels, reported by Prasad et al. (1961), was a patient with vitamin-D-resistant rickets before treatment.
Administration of magnesium to two children who had rickets, hypocalcemia, and high levels of alkaline phosphatase, despite very high doses of vitamin D2, corrected the biochemical abnormalities and produced X-ray evidence of bone healing (V. Reddy and Sivakumar, 1974). These investigators reported a 5-year-old boy and a 2-year-old girl with rickets, whose hypocalcemia and serum alkaline phosphatase levels of 24.1 and 42.6 Bodansky units failed to respond to several doses of 600,000 IU of vitamin D2. In the boy, serum alkaline phosphatase levels rose further following the high-dosage vitamin D therapy. Severe hypomagnesemia (0.4 mEq/ liter) was then detected and oral magnesium supplementation (10 mEq/day as MgCl2 was started. All biochemical determinants became normal within 4 weeks. The serum magnesium level of the baby girl, who had received 4,000 IU of vitamin D from early infancy, was found to be 0.6 mEq/liter on admission. She was given 600,000 IU of vitamin D daily for 10 days without biochemical improvement. Magnesium supplementation resulted in prompt fall of high levels of serum phosphatase activity, and rises in serum magnesium, calcium, and phosphorus. She was not given the prescribed magnesium at home, and within a month her biochemical abnormalities had recurred. They were promptly corrected on reinstitution of magnesium therapy (Fig. 12-5). Since the diets of these children were not deficient in magnesium, the investigators believe they are probably magnesium malabsorbers.
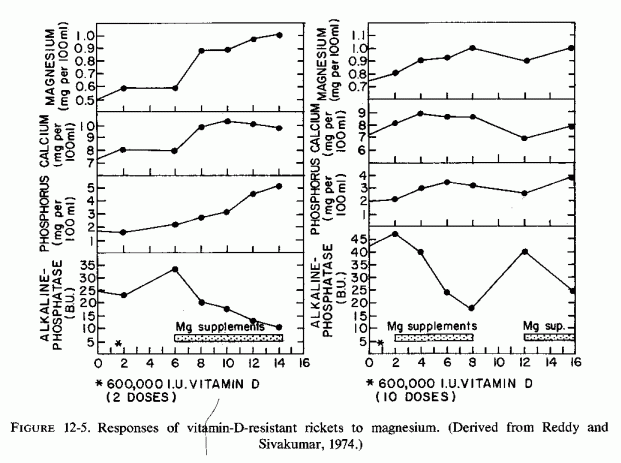{kind=link}
Rapado et al. (1975) termed the rickets of their 12-year-old patient "magnesium-deficient rickets." She had a long history of polyuria and was found to have nephrocalcinosis with persistent hypercalciuria. After 7 months of treatment with sodium cellulose phosphate (10g/d), her hypercalciuria was corrected, but she developed hypocalcemia, and increased serum alkaline phosphatase. Treatment was then changed to hydrochlorothiazide for two months, after which she developed tetany and osseous pain. Her serum calcium was then 6.9 mg/100 ml, her serum magnesium was 0.5 mEq/liter and her urinary outputs of magnesium and calcium were subnormal. She exhibited subnormal response to PTH. By this time she had signs of overt rickets in wrists and knees. Intramuscular magnesium supplementation (1.5 g/day) for a month resulted in disappearance of the radiologic signs of rickets and correction of the hypomagnesemia and hypocalcemia. On readmission six weeks later, her serum magnesium was again low (1 mEq/liter). On a normal magnesium intake (336 mg she absorbed only 0.2%; thus she represented another instance of magnesium malabsorption. Rapado and Castrillo (1976/ 1980a) have identified another patient with magnesium-dependent rickets, nephrocalcinosis, and who has magnesium malabsorption. Rapado et al. (1975) recommend that patients with vitamin-D-resistant nephrocalcinosis, or failure in response to PTH be evaluated for magnesium deficiency.
Patients with steatorrhea, enteritis, or bypass surgery for obesity have exhibited vitamin-D-refractory osteomalacia (Blackard et al., 1962; Prost et al., 1972; Reitz and Weinstein, 1973; Medalle et al., 1976). Although hypocalcemia is more frequently reported in this disorder, and the hypophosphatemia is commonly attributed to resultant secondary hyperparathyroidism, magnesium deficiency is also common. Reversal of vitamin D resistance has been produced in such patients with magnesium repletion (Medalle et al., 1976).
Although the magnesium status has been shown to influence the response to vitamin D in animals (magnesium deficiency increasing vitamin D requirements) and magnesium is a cofactor in vitamin D conversion to its active steroid-metabolites, its role in vitamin D metabolism in clinical magnesium depletion is not clear. For example, patients who were hypomagnesemic as a result of malabsorption synthesized no less 1,25-(OH)2D3 than did normomagnesemic, hypocalcemic, vitamin D-deficient patients (Lukert, 1976/1980). The active vitamin-D-derived hormones are necessary both for normal intestinal absorption of calcium and for bone calcium turnover (Reviews: Norman, 1974, 1977; DeLuca, 1976). The evidence that vitamin D is necessary for intestinal absorption of magnesium and lowers bone magnesium levels suggests that the active metabolites must also influence magnesium metabolism, and abnormality in vitamin D metabolism probably influences bone integrity as a function, not only of changes in handling of calcium but of magnesium. Avioli et al. (1967) found increased levels of vitamin D metabolites without calcium absorptive activity in hypophosphatemic rickets. How such metabolites influence magnesium absorption or bone levels has not been reported.
Vitamin D increases renal tubular reabsorption of phosphorus (Harrison and Harrison, 1941). A vitamin-D-dependent intestinal phosphate-transport mechanism (DeLuca, 1976) can partially explain the hypophosphatemia of vitamin-D-refractory rickets. Association of abnormal vitamin D metabolism with secondary hyperparathyroidism (Arnaud et al., 1970) supports the premise that hyperparathyroidism, whether secondary to intestinal malabsorption or to abnormal vitamin D metabolism, contributes to impaired phosphate reabsorption by the kidneys. Clinical evidence of the importance of the abnormality of vitamin D metabolism in hypophosphatemic rickets has been obtained by demonstration of response of patients with this disorder to the 25-(OH)D3 (Rosen and Finberg, 1972, 1973), to the 1,25-(OH)2D3 (Fraser et al., 1973; Avioli and Haddad, 1973; Balsan et al., 1975) and to 1,a(OH)D3 (Balsan et al., 1975; Rosen and Finberg, 1975/1977). The active metabolites, like the parent substance, vitamin D3, correct the malabsorption of calcium and the impaired bone mineralization, but do not influence the form of the disease that is characterized by intrinsic defective renal tubular phosphate reabsorption (Brickman et al., 1973; Glorieux et al., 1973). Possibly relevant to this partial failure of treatment is the demonstration of phosphaturia in magnesium-deficient rats (despite their hypercalcemia, and even in those that were parathyroidectomized) by Ginn and Shanbour (1967).
A paradoxical facet to vitamin D resistance is that, in addition to its association with hyperparathyroidism and hypophosphatemia, magnesium-reversible vitamin D refractoriness has long been recognized in hypoparathyroidism with hyperphosphatemia (Homer, 1961; K. Jones and Fourman, 1966; Harrison et al., 1967). A child with idiopathic hypoparathyroidism who had hypocalcemia and low serum alkaline phosphatase levels responded to PTH with phosphaturia, but without correction of her hypocalcemia (Rösler and Rabinowitz, 1973). She was then given up to 600,000 IU of vitamin D and 3-4 g of calcium lactate for 13 weeks, and then dihydrotachysterol, without raising her blood calcium level. Her serum magnesium was then found to be 0.5 mEq/liter, and she developed convulsions and tetany. Magnesium repletion produced rapid improvement. Medalle and Waterhouse (1973) reported a biochemical picture of hypoparathyroidism, hypocalcemia and hyperphosphatemia in a patient with severe magnesium depletion of chronic alcoholism, and suggested that magnesium deficiency be considered in the differential diagnosis of hypoparathyroidism, pseudohypoparathyroidism, and renal failure. Their patient did not exhibit a normal phosphaturic response to PTH while she was magnesium depleted. Magnesium therapy corrected her hyperphosphatemia promptly; correction of her hypocalcemia was more gradual. This finding suggested to the investigators that hyperphosphatemia does not occur unless the magnesium depletion is severe. They noted that it occurs less frequently than does hypocalcemia in magnesium-deficient patients, and has been reported with selective experimental magnesium deficiency (Shils, 1969a) or with isolated magnesium malabsorption (Dooling and Stem, 1967; Skyberg et al., 1967; 1968; Stromme et al., 1969; Nordio et al., 1971).
It might be well, also, to ascertain whether the high vitamin D requirements of low-birth-weight infants (who are also subject to hypoparathyroidism and hyperphosphatemia) might be contributed to by magnesium inadequacy. Vitamin-D-resistant rickets of biliary atresia, which is responsive to 25-OHD3 (Daum et al., 1976; Rosen and Finberg, 1975/1977), might also be responsive to magnesium, magnesium deficiency having been demonstrated in such infants (A. Kobayashi et al., 1967, 1974). On the other hand, a defect in intestinal absorption of magnesium, as well as of calcium, has been found in idiopathic hypoparathyroidism, which improved following treatment with 25-OHD3 (Rosen and Finberg, 1975/1977).
Magnesium deficiency affects parathyroid hormone (PTH) secretion, and target organ response, and several syndromes are associated primarily with skeletal abnormalities that might have abnormal magnesium metabolism as a common denominator. For example, idiopathic hypoparathyroidism and pseudohypoparathyroidism and variations of these disorders that have been given cumbersome names (e.g., pseudopseudohypoparathyroidism, pseudohypohyperparathyroidism) might be explicable on the basis of different phases and degrees of magnesium deficiency and its influence on response to calcemic or hypocalcemic therapy. Bronsky (1970) criticized the nomenclature used for the variations in disorders that are associated with Albright's osteodystrophy (brachydactyly, stocky body, and round face) or dyschondroplasia with soft tissue calcinosis. Selected from his tabulated forms of parathyroid disease are several that might derive from abnormalities of magnesium metabolism, or from maternal, perinatal, or later dietary imbalances (Table 12-1). Patients with osseous or soft tissue calcinosis frequently have close relatives with parathyroid disease or convulsive disorders, which is suggestive of a possible contributory magnesium deficit. Bronsky (1970) stressed the relationship of steatorrhea to parathyroid disease (both hypersecretion and resistant hypoparathyroidism), an observation that supports the concept of underlying magnesium deficiency. Discussed earlier are gestational hyperparathyroidism and neonatal hypoparathyroidism and their likely interrelationships with magnesium deficiency.
{kind=link}
Albright et al. (1942), who first described the pseudohypoparathyroid dyschondroplasia syndrome, noted that patients with this disorder had convulsions themselves and had siblings with epilepsy, were mentally retarded, and were resistant to high doses of calcemic agents. These manifestations, and the histories of infantile respiratory distress (reported in the original and subsequent cases) resemble those of infants who developed cardiovascular and/or renal abnormalities that are speculated also to be related to magnesium deficiency. The patient reported by C. Lowe et al. (1950), who had an early symptom-complex very much like infants with hyperreactivity to vitamin D, early developed signs of vitamin-D-resistant rickets and spontaneous fractures, such as are seen in hypophosphatasia (related to magnesium deficiency). When she was found to have steatorrhea, the authors speculated that malabsorption might have contributed to her disorder; the malabsorption certainly could have caused magnesium depletion. Talbot et al. (1954) described the syndrome in twins, and commented that hypocalcemic symptoms of this disorder usually date from the neonatal period, further suggestive evidence of magnesium deficiency.
An infant who died at two months of age, who had neonatal hypoparathyroid, hypocalcemia, and hypomagnesemia, had had similar signs, persistent diarrhea, and renal tubular acidosis, but no X-ray evidence of rickets or soft tissue calcinosis. PTH injection evoked a phosphaturic and calcemic response, but lowered her serum magnesium to subnormal levels. Calcium therapy further lowered it, and caused hypercalcemia (Taitz et al., 1966). At autopsy, neither parathyroid nor thymic tissue was found, and there were intraluminal renal tubular calcium deposits. The bones were not examined. Whether this case reflects profound prenatal parathyroid suppression, or a genetic defect, complicated by a metabolic disorder as proposed by the investigators, and whether longer survival would have resulted in overt skeletal abnormalities, is not possible to aver.
The manifestations of the disorders categorized as "idiopathic" hypoparathyroidism and pseudohypoparathyroidism or dyschondroplasia, predominantly on the basis of presence or absence of phosphaturic response to PTH, were compiled by Bronsky et al. (1958). Their tabulation of symptoms and signs of patients diagnosed as "idiopathic" or "pseudohypoparathyroid" (Table 12-2)and their comment that both are idiopathic, the cause not being known, is valuable. It should be noted, however, that current emphasis is on failure of both kidneys and bone of pseudohypoparathyroid patients to respond to PTH, and on failure of 3',5' cyclic adenine monophosphate (cAMP) response (Chase et al., 1967, 1969; Drezner et al., 1973). That this abnormality might underlie the refractoriness of bones and kidneys to PTH (Coburn et al., 1972) points toward a possible underlying magnesium deficiency, the synthesis of cAMP having an absolute magnesium requirement (Sutherland et al., 1968).
Evidence has been presented that prenatal magnesium deficiency can contribute to neonatal hypoparathyroidism, and that subsequent dietary imbalances might predispose to permanent damage. Not only the cardiovascular system, but the skeletal and renal systems can be affected, the manifestations depending on combinations of genetic metabolic abnormalities, dietary imbalances, and treatment. Perhaps the higher incidence of thickened calvaria and the much higher incidence of soft tissue calcinosis and mental retardation of the pseudohypoparathyroid group than of the "idiopathic" group might reflect a (postulated) greater magnesium deficit and a consequent need for higher doses of calcemic agents to correct hypocalcemia, with resultant signs resembling those of infantile hypercalcemia. The coarse trabeculation of the short bones of this group of patients, in fact, resembles that seen in experimental magnesium deficiency. Also, the occasional exostosis or tumorlike growth on long bones resembles that seen in patients with hypophosphatasia or osteogenesis imperfecta (speculated to be contributed to by magnesium deficiency) and seen in experimental magnesium deficiency. Slipped epiphyses, such as have been reported in a few patients with magnesium deficiency and that are in accord with the epiphyseal abnormalities of experimental magnesium deficiency have also been reported in different forms of pseudohypoparathyroidism (Bronsky et al., 1958; Frame et al., 1972). Young patients with the disorder, termed "pseudohypohyperparathyroidism," who had osteosclerosis in the skull, osteitis fibrosa in long and flat bones, and slipped epiphyses, have had serum magnesium levels reported within normal limits (Frame et al., 1972). The adolescent girl, who had had resistant rickets and subsequent nephrocalcinosis from infancy, and who developed osteitis fibrosa cystica and parathyroid adenomas after years of high-dosage calcemic therapy, had both hypercalcemia and hypermagnesemia on admission to the hospital (W. Thomas and Fry, 1970). The past history of nephrocalcinosis should predispose to renal magnesium wastage, which either did not exist in this patient or was masked by PTH-mobilization of bone minerals preoperatively. More intensive studies of the magnesium status of such patients are necessary to clarify whether cellular magnesium deficit might exist, despite lack of hypomagnesemia. Studies of magnesium metabolism in members of families with either hypo- or hyperparathyroidism that has a genetic component (whether the complete syndrome exists, or only some of the manifestations) might be fruitful.
The condition termed "renal rickets" has long been known to be associated with severe skeletal distortions, acidosis, renal calcinosis, hyperparathyroidism, and mental and growth retardation (Shelling and Remsen, 1935; Price and Davie, 1937). The child reported by Price and Davie (1937) is of particular interest in building up a case for primary magnesium deficiency, since he was the product of the seventh pregnancy, and had been born the year after a miscarriage and the year before two additional miscarriages. As has been discussed, frequent pregnancies are likely to predispose to fetal magnesium deficiency and to spontaneous abortions. This child had generalized osteoporosis and alternate sclerosis and rarefaction of the skull at the age of 14, florid rachitic changes at the extremities, slipped epiphyses, renal damage, deafness, and evidence of mental retardation. At autopsy, it was found that the radiologic diagnosis of slipped femoral epiphysis was incorrect; he actually had collapse of the metaphysis of the neck of the femur, and bone was replaced by a mixture of fibrous tissue and cartilage. All four of his parathyroids were hyperplastic. There were numerous small foci of calcification in his kidneys. These investigators question whether it is necessary to be certain that the renal lesion has preceded the other findings for a diagnosis of renal rickets to be made, as was the case in the similar boy reported by Shelling and Remsen (1935). That boy had hypercholesterolemia, hypertension and arteriosclerosis, and hyperphosphatemia despite parathyroid hyperplasia, elevated PTH levels, and skeletorenal lesions much like those of the child reported by Price and Davie (1937).
In 1927, L. Parsons described five children with fragile bones and rickets secondary to celiac disease. He noted that the skeletal deformities usually do not develop until the age of seven years. One of his patients had blue sclerae similar to that seen in osteogenesis imperfecta, which gradually became normal in color as the malabsorption improved. Spontaneous pseudofractures were sometimes seen, severe osteoporosis, and persistently fragile bones, even after control of the malabsorption, and despite treatment with cod liver oil. These manifestations are of interest because of their similarity to those of experimental magnesium deficiency and to diseases speculated to be contributed to by magnesium depletion. Prost et al. (1972) have described osteomalacia secondary to malabsorption in adults. They correlated osteomalacia and pseudofractures with hypomagnesemia in two instances, and recommended evaluation of the magnesium status with a view to its repair, in an effort to restore vitamin D responsiveness in such patients.
12.4.4.1. High Vitamin D and Calcium/Low Magnesium
Skeletal changes similar to those seen in magnesium-deficient animals given diets relatively high in calcium and vitamin D, or in hypervitaminosis D studies, are seen in clinical osteopetrosis. Storey (1960) has reviewed the X-ray, histological, and biochemical findings of this disease. Pathognomonic is alternation of radiopaque and radiolucent transverse bands running parallel to the epiphyseal cartilage of the long bones, and to the surface of other bones. Histological studies have shown that the bone is increased in amount, but abnormal. There is some normal bone, islands of densely calcified cartilage near the epiphyses, and areas of osteoid tissue so poorly calcified as to resemble rickets. Microradiographs show areas of high and low bone density, and exaggerated thick radiopaque "cementing" lines on the surface and concentrically around immature Haversian systems. Intense bone resorption is also occasionally seen. These changes are often accompanied by generalized calcinosis of arteries, kidneys, ligaments and tendons, and other soft tissues. Biochemical changes are inconstant, depending on the stage of the disease. Serum calcium levels are usually normal, but hypercalcemia has been reported. Serum phosphorus is often low, with a Ca X P product suggestive of vitamin-D deficient rickets, or sometimes of vitamin-D-refractory rickets. Storey (1960) confirmed the bone changes of "hypervitaminosis D rickets" in rats, as described by Ham and Lewis (1934), who found flattened, thinned epiphyses, numerous thickened trabeculae, and matrix ranging from normal to poorly calcified, and then possibly "compensatory" excess osteoid. When he gave high doses of vitamin D intermittently to rats (Storey, 1960), the bone changes were very much like those seen in clinical osteopetrosis. The base of the skull became extremely dense and thick. Storey (1960), puzzled over the bone changes caused by excess vitamin D in his own and other studies and considered mediation by hormonal responses, calcium, phosphorus, and "as yet unelucidated systemic disturbances." The similarity of the excess vitamin-D-induced changes of the epiphyses, trabeculae, and matrix, to those described by Bernick and Hungerford (1965) in magnesium-deficient rats suggests that magnesium loss from bone, caused by excess vitamin D, might be a contributory factor.
Magnesium balance data were obtained in a series of balance studies (done over an eight-month period) in an infant with roentgenologic evidence of osteopetrosis but with biochemical evidence of hypophosphatemic rickets: marginal hypocalcemia, hypophosphatemia, and elevated serum alkaline phosphatase (Pincus et al., 1947). Although the authors did not comment on the magnesium findings, analysis of their data shows that in the preliminary test period (at two and one-half months of age) the baby retained 10 times as much calcium as magnesium, and her calcium/magnesium absorptive ratio was 7/1. Two months after her vitamin D2 supplementation was increased severalfold over the usual dose, her retention of calcium was 16 times that of magnesium, and she absorbed 10 times as much calcium from the gut. During the last balance period (at 10 months of age) when her magnesium intake had been increased to 553 mg per day and her calcium intake had also been increased but proportionally less, the ratio of intestinal calcium to magnesium was 6/1. Her retention of Ca/Mg, however, was 9/1, a greater percentage of the absorbed magnesium being excreted in the urine. Her serum alkaline phosphatase had fallen by that time to hypophosphatasia-levels, her serum calcium remained marginally low, but her serum phosphorus had risen to 5.5 mg/100 ml. A trial of parathyroid extract transiently increased the serum calcium to within normal limits, and the serum phosphorus gradually fell to 3 mg. After 6 weeks, the child became refractory to parathyroid treatment. She died at 16 months, and her osteopetrosis was confirmed at autopsy
Infantile hypercalcemia is associated with osteosclerosis. The first such patient reported was a dwarfed infant with hypercalcemia, cardiovascular and renal calcinosis, and mental retardation (Lightwood, 1932). What was then a rare syndrome appeared much more commonly in the literature in the 1950s, during an era of excessive fortification of milk with vitamin D in the British Isles (Review: Seelig, 1969). The skeletal abnormalities were less commonly reported than was the severe cardiovascular, renal, and mental damage, which was termed the supravalvular aortic stenosis syndrome (SASS, Editorial, Br Med J, 1956). Fanconi and Girardet (1952) described an infant with the full syndrome. British babies were then reported with radiographic evidence of excessive deposition of sclerotic bone at the base of the skull, in periorbital bones, at ends of long bones, and at the borders of the vertebrae (Creery, 1953; Russell et al., 1954; Dawson et al., 1954; Lowe et al., 1954; Stapleton and Evans, 1955; Schlesinger et al., 1956; Joseph and Parrott, 1958). The amount of vitamin D estimated to be consumed by the affected children ranged from 1000 to 3200 IU, an amount that is not infrequently provided by the American diet. And, in fact, these lesions have not been limited to the British babies. The syndrome has been described in continental Europe and in America, the cardiovascular anomalies more frequently, the skeletal changes less frequently. Shiers et al. (1957) reported four children from one-and-a-half to almost five years of age, all of whom had roentgenologic evidence of osteosclerosis and other signs of hypervitaminosis D, but none of whom had histories of its excessive consumption. One had multiple bands of sclerosis parallel to the growing ends of the long bones, and distorted shafts; one had increased skull density, particularly at the base, with increased density of vertebral and carpal bones and of epiphyses, and one had rachitic-like lesions of the ends of the long bones but generalized osteosclerosis. The authors noted that the most heavily sclerosed bone had been laid down in utero. The oldest child, who was also hypothyroid, had very heavy osteosclerosis, particularly in the cranial and facial bones. All bones were affected, with bands of varying density. Three infants, who had been born prematurely, developed the classic signs of severe hypercalcemia by 6 months of age, and were found to have osteosclerosis at 10 to 17 months of age (Singleton, 1957; Daeschner and Daeschner, 1957; Snyder, 1958). None had been given more than 1000 IU of vitamin D as supplements (in addition to that provided by milk and other fortified foods). A Swiss child of low birth weight was born to a mother who later developed diabetes mellitus (a condition associated with low magnesium levels) and developed the full-blown syndrome by 5½ months of age after high-dosage vitamin D (Illig and Prader, 1959) Another infant who was small at birth, born to a mother who had taken 1000 IU vitamin D daily during much of her pregnancy, developed the syndrome at 4 months of age (Fraser et al., 1966). Others, who developed the classic signs of hypercalcemia, SASS, and osteosclerosis at 9 to 18 months of age, were normal-sized at birth and had not been given high-dosage vitamin D supplements (O'Brien et al., 1960; N. David et al., 1962; Garcia et al., 1964; D. Fraser et al., 1966). The youngest infant with hypercalcemia and osteosclerosis had not had high-dosage vitamin D but had been given supplemental calcium (Wilkerson, 1964). Hyperreactivity to vitamin D is suspected in these children.
Infants and children have developed the complete hypercalcemic syndrome, including osteosclerosis, after massive intermittent doses of vitamin D (Amann, 1959; Manios and Antener, 1966). A child who had received excessive daily vitamin D supplements from his third through fifth years of age developed periarticular calcification and hypertension as well as osteosclerosis. He died, two years after his excessive supplements had been stopped, with renal failure and coronary atherosclerosis (DeWind, 1961).
Search for possible prenatal factors in the pathogenesis of infantile hypercalcemia and the SASS, led to studies of pregnant rabbits overdosed with vitamin D. W. Friedman and Mills (1969) found that some of the young had premature closure of the cranial bones, osteosclerosis, and palatal abnormalities similar to those seen in infants and children with infantile hypercalcemia and the SASS. Rowe and Cooke, (1969), considering the role of maternal vitamin D in the genesis of the excessive fetal mineralization in the rabbits (W. Friedman and Mills, 1969; W. Friedman, 1968), commented that mothers of children with the SASS had not usually had histories of vitamin D overdosage during pregnancy. They noted that Friedman and Mills (1969) had considered the possibility of acquired decreased tolerance of vitamin D. They suggested that an infant who had undergone excessive mineralization in utero might be unduly susceptible to both hypercalcemia and osteopetrosis thereafter. It should be noted, here, that early studies of the effects of supplementing pregnant women with only moderate doses of vitamin D showed that the fetuses tended to have narrower cranial sutures and greater bone density than did the fetuses of control nonvitamin-D-supplemented mothers (Finola et al., 1937; Brehm, 1937; Review: Seelig, 1978). Rowe and Cooke (1969) proposed that there might be a failure of regulating mechanisms for blood calcium in infants with SASS and osteosclerosis, and that there is probably a multifactorial basis for the difference in susceptibility to the disease. A factor that should be considered is the possible role of magnesium deficiency: gestational, magnesium malabsorption, or vitamin D induced. The ranges of susceptibility to vitamin D toxicity (Fanconi, 1956), and the magnesium loss caused by excess vitamin D should also be taken into account.
Intermittent magnesium treatment of the constipation characteristic of infantile hypercalcemia has been mentioned by some of the investigators of infantile hypercalcemia (Creery, 1953; Lowe et al., 1954; Forfar, thesis). Stapleton and Evans (1955) noted that a hypercalcemic infant fed a formula free of calcium and magnesium exhibited a steady drop in serum magnesium levels (to 1.4 mEq/liter). Lowe et al. (1954) reported hypomagnesemia in a mild case and hypermagnesemia in a severe case. Metabolic balance studies of severely hypercalcemic infants showed that they were in magnesium equilibrium (McDonald and Stapleton, 1955), only slightly positive (+ 1.3 mg/kg/day) or negative (Forfar, thesis). Fellers and Schwartz (1958), who studied two infants with severe hypercalcemia even when all vitamin D was removed from the diet, and who suggested that the disease is caused by abnormal vitamin D metabolism (Fellers and Schwartz, 1958b), reported that when calcium and vitamin D were deleted from the diet, the children went into strongly positive magnesium balance. These data suggest that magnesium deficiency may be part of this syndrome since infants should be in strongly positive magnesium balance (Seelig, 1964, 1971). Dalderup (1960) was the first to propose that magnesium deficiency might be contributory to this disorder.
The cited metabolic balance study by Pincus et al. (1947) supports the premise that magnesium malabsorption might be an initiating disorder that might contribute to hypophosphatemic rickets. Vitamin D, given to infants whose bone matrix is abnormal because of magnesium deficiency, might lead to hypermineralization, such as is produced in rats on high-dosage vitamin D plus calcium. The development of hypophosphatasia after 8 months of high-dosage vitamin D in the infant studied by Pincus et al. (1947), and the hypophosphatasia found in infantile hypercalcemia with hyperreactivity to vitamin D, suggest that intensification of magnesium deficiency by excessive vitamin D might be at fault, alkaline hypophosphatasia also being characteristic of magnesium deficiency.
However, once hypercalcemia is part of the clinical syndrome, it should be corrected before attempting to correct the magnesium deficiency with a parenteral magnesium load. Alkaline and pyrophosphatases (which destroy the calcification-inhibiting polyphosphates and pyrophosphates) are found, not only in bone but in the kidneys, cardiovascular, and other soft tissues. Since the phosphatases are magnesium dependent, administration of magnesium (in the face of hypercalcemia) might increase the risk of metastatic calcification, as had been suspected by the physicians who treated hypercalcemic infants (supra vide). Whittier and Freeman (1971) have provided experimental evidence that administration of magnesium to rats made hypercalcemic by hypervitaminosis D did in fact increase renal and myocardial calcification.
Congenital osteopetrosis need not be associated, however, with hypercalcemia. Rosen and Haymovits (1972) have reviewed the evidence that the disease is characterized by impaired bone resorption, and have speculated that a defect in lysosomal functions might be a significant factor in its pathogenesis. They demonstrated increased levels of the hepatic lysosomal enzyme, β-glycerophosphatase (the significance of which is unclear), and increased frequency of hepatic electron-dense mitochondrial particles. Whether these granules are comparable to those reported in myocardial mitochondria in magnesium deficiency and whether they are an indication of magnesium deficiency is speculative.
In considering the effect of vitamin D and calcium supplementation to pregnant women, the active transport of calcium across the placental barrier and the effect of high calcium levels on calcitonin (CT) secretion should also be taken into account. Acute hypercalcemia (in rats) has lowered the CT content of thyroid C cells (Gittes et al.., 1968), and has increased plasma immunoreactive CT levels in several species of animals (Littledike et al., 1972). There is direct evidence that the hypercalcemia caused by excessive vitamin D (in cows) increases CT release (Young and Capen, 1970). In the gray lethal mouse, which develops osteopetrosis, it has been proposed that the primary lesion is hyperplasia of thyroid C cells, with overproduction of CT (D. Walker, 1965, 1966). There is evidence that CT not only inhibits bone resorption (Johnston and Deiss, 1966; Bélanger and Rasmussen, 1968; Raisz et at., 1968; Baylink et al., 1969; Hirsch and Munson, 1969), but that it also increases bone calcification, growth, and repair (Wase et al., 1967; Pallasch, 1968; Ziegler and Delling, 1969; Delling et al., 1970; Gaillard and Thesingh, 1968; Matthews et al., 1972; Salomon et al., 1973). Fetuses infused with calcium secrete CT (Littledike et al., 1972; Garel et al., 1973, 1974, 1976; Garel and Barlet, 1974) and the high fetal and cord CT levels are presumed to play an important role in normal bone growth and calcification (Samaan et al., 1973, 1975). Thus, it seems likely that hypercalcemia of fetuses of mothers given excessive vitamin D might cause abnormally high fetal CT levels and increase bone mineralization. It is possible that low fetal magnesium levels, such as is postulated to be not uncommon, also increases CT secretion. The influence of the fetal magnesium/calcium ratios on the PTH/CT responses will influence the nature of the changes induced in fetal and infantile bone.
12.5.1. Osteogenesis Imperfecta
The similarity of the bone lesions in young of rats given excessive vitamin D during pregnancy to those of osteogenesis imperfecta, the magnesium depletion caused by hypervitaminosis D, and the infantile osteopenia and spontaneous fractures seen in infants likely to have magnesium deficiency (supra vide), suggest that the magnesium status of members of families with osteogenesis imperfecta be explored. It is conceivable that familial malabsorption or renal wastage of magnesium might be contributory to the familial occurrence of osteogenesis imperfecta.
Whether osteogenesis imperfecta is a separate entity from the severe early form of hypophosphatasia, the bone lesions of which are indistinguishable from it, is not yet certain. The essential difference is in the serum phosphatase levels that have been reported. Hansen (1934) confirmed earlier reports that patients with osteogenesis imperfecta do not have the low serum alkaline phosphatase levels that are characteristic of hypophosphatasia. However, he analyzed tissues of a child who died of the disease, without having received unusual medication, and found almost complete absence of phosphatase in the periosteum and subperiosteal structures, where it is normally abundant. Solomons and Styner (1969) studied 28 patients (2 days to 14 years of life) with this disease and found the collagen biopsies completely prevented mineralization at pH 7.4, and that pyrophosphatase in the presence of magnesium (3 X10-3) markedly reduced the inhibition. Addition of magnesium without the enzyme partially reduced the inhibition of mineralization. They reported that bone from patients with osteogenesis imperfecta had much higher levels of pyrophosphate than did normal bone. This excessive pyrophosphate could be almost completely removed by in vitro treatment with pyrophosphatase plus magnesium. They also reported significantly higher than normal serum pyrophosphate levels in serum and urine, a finding not corroborated by R. Russell et al. (1971), who found higher than normal plasma levels only in hypophosphatasia. (The latter investigators, however, cautioned that plasma pyrophosphate levels might not be in equilibrium with that in bone or other tissues.) In view of the difference in pyrophosphate levels reported by the two groups, it is not possible to evaluate the significance of Solomons' and Styner's (1969) clinical report that administration of magnesium salts (2-6 mg/kg) to four patients with osteogenesis imperfecta lowered their serum and urine pyrophosphate levels toward the normal range.
J. Albright and Grunt (1971) studied magnesium balance (among other elements) in five children with osteogenesis imperfecta before and after fluoride treat ment. All had negative magnesium balances, which were not affected by the fluoride. Riley and Jowsey (1973) treated three patients with magnesium oxide (15 mg/ kg/24 hr) with only minor changes in bone formation and resorption, as measured by microradiography of iliac crest biopsies (Table 12-3). Whether the slight increases in bone formation and increases in bone formation and resorption noted in the two children with the severe form of the disease might have continued with prolongation of magnesium therapy would have been of interest. The older child, whose disease was less severe, and whose microradiographic studies showed less abnormal bone turnover, showed a drop in bone resorption (but still to within normal limits) but also a decrease in new bone formation. Intestinal and renal handling of magnesium should be correlated with bone response.
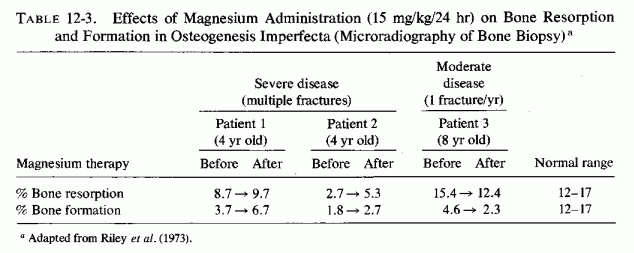{kind=link}
Benign hyperplastic callus formation, which simulates osteosarcoma, has been reported in patients with osteogenesis imperfecta. Banta et al. (1971) reviewed 21 published cases, and 2 of their own, of such superabundant callus (usually of the tibia or femur but sometimes of the pelvis) that led to amputation for sarcoma in several instances. Replacement of muscle tissue by the extensive fracture callus was consistent with myositis ossificans. One of their patients (a young man of 22) also had bilateral dislocation of the radial heads and ankylosis of the spine. These abnormalities are noted because of the demonstration of exuberant growth of the femur, simulating osteosarcoma, of magnesium-deficient rats, and of the possibility that slipped epiphyses and chondrocalcinosis, including spondylitis, might be related to magnesium deficiency. Further evidence of abnormalities in collagen of patients with osteogenesis imperfecta derives from studies of skin collagen (Haebara et al., 1969; C. Stevenson et al., 1970; R. Smith et al., 1975) and bone collagen and matrix proteins (Haebara et al., 1969; Dickson et al., 1975). Thin scleral collagen has been suggested as a factor in the characteristic blue sclerae. If the abnormality in bone matrix is similar to that produced by experimental magnesium deficiency (Bernick and Hungerford, 1965; Trowbridge and Seltzer, 1967), and if the propositus and his close relatives can be shown to absorb or retain magnesium abnormally, we might have another clue to the pathogenesis of this disease.
Another fragment of evidence that magnesium deficiency might be participatory is the aminoaciduria detected in some patients with osteogenesis imperfecta and in members of their families (Chowers et al., 1962; Brigham and Tourtelotte, 1962; Summer and Patton, 1968). Five children with osteogenesis imperfecta were born to three families, almost all the members of which had aminoaciduria (Chowers et al., 1962). The authors had investigated the amino acid excretory patterns of the families because of the frequent association between bone-wasting diseases and renal tubular dysfunction (e.g., osteomalacia, rickets, Fanconi syndrome, and hyperparathyroidism). Aminoaciduria has been produced in animals by experimental magnesium deficiency and is seen in patients with hyperreactivity to vitamin D (Fanconi and Girardet, 1952) or with intestinal malabsorption (Muldowney et al., 1968), both conditions in which magnesium deficiency is demonstrable or suspected. Abnormal amino acid urinary output has been repeatedly demonstrated (Seelig and Berger, unpublished observation) in a woman with rapidly progressive osteoporosis, latent tetany of magnesium deficiency (Seelig et al., 1975) and renal magnesium wastage (Seelig et al., 1976/1980). The amino acid urinary excretory pattern of infants who have been given excessive vitamin D or who have hyperreactivity to vitamin D has rarely been reported. Drummond et al.(1964), however, ascertained that infants with familial hypercalcemia and nephrocalcinosis have abnormal tryptophan metabolism, termed the "blue diaper syndrome." This abnormality is of interest, since comparable abnormal metabolites of tryptophan are excreted in vitamin B6 deficiency or abnormality, and pyridoxine enzymes are magnesium dependent (Review: Durlach, 1969b)
Osteogenesis imperfecta, like hypophosphatasia, abnormalities in vitamin D or magnesium metabolism, and congenital heart diseases that have been correlated with either or both of these metabolic abnormalities, can be isolated or familial. It is of interest that the bone and cardiac disorders have been seen in the same patient, sometimes in association with renal calcinosis. For example, Coleman (1959) reported a baby with osteogenesis imperfecta, who died with nephrocalcinosis and thrombosis, among a series of 24 with infantile hypercalcemia, whose ECG changes (ST-T abnormalities) were not related to serum calcium levels. Examination of the ECG data shows similarities to those reported in conditions associated with magnesium deficiency (Review: Seelig, 1969a). It has been suggested that idiopathic hypertrophic subaortic stenosis might similarly be associated with hypercalcemia (McFarland et al., 1978). Whether the growth retardation and skeletal abnormalities (particularly of the face and base of skull, leading to cardiofacies, and of the chest) that have been seen in cardiac outflow abnormalities (Chapter 4) are similarly mediated cannot be averred. Investigation of the metabolism of magnesium and of vitamin D of the propositus, and especially of infant siblings and mother, might provide insight into the etiology of these forms of combined cardiac and skeletal abnormalities.
It should be recalled that infantile hypercalcemia is frequently associated with the supravalvular aortic stenosis syndrome and with other cardiac outflow abnormalities. It is thus provocative that osteogenesis imperfecta has been reported in patients with aortic coarctation (Remigio and Grinvalsky, 1970) and in patients with valvular abnormalities requiring correction by open heart surgery (Criscitiello et al., 1965; Heppner et al., 1973; Wood et al., 1973; Waters et al., 1977). Perhaps most directly suggestive of the role of gestational magnesium deficiency in the pathogenesis of the combined congenital abnormalities of osteogenesis imperfecta, valvular disease, and aortic coarctation, are the two infants born with these disorders to a young woman who had had multiple pregnancies at short intervals (Remigio and Grinvalsky, 1970). They were the products of her ninth and tenth pregnancies, the seventh and eighth having terminated as spontaneous abortions. Such frequent pregnancies have been shown to be associated with maternal magnesium depletion. However, there might well have been a genetic predisposition to skeletal abnormalities, since the first two siblings had abnormalities of their hips. McKusick (1966) and Shoenfeld et al. (1975) have cited premature arteriosclerosis in osteogenesis imperfecta, another hint at possible underlying magnesium deficiency that is probably caused by defective ability to absorb or retain magnesium.
Hyperparathyroidism has also been associated with magnesium loss, and thus the coexistence of hyperparathyroidism and osteogenesis imperfecta tarda in women in their late forties or early fifties (Goldzieher et al., 1957; Quay et al., 1968; Salti et al., 1973; Woolfson et al., 1975) provides still another piece of circumstantial evidence linking magnesium deficiency with this form of osteopenia. Whether decreased estrogen secretion, which antagonizes parathyroid hormone activity, allows for an occult disorder to become overt in patients with mild forms of this disease is speculative.
Patients with osteopenias are commonly treated with high-dosage calcemic agents, which increase both magnesium loss and extraskeletal calcification. Thus, the combination of bone defects with damage to such organs as the heart, arteries, and kidneys, and ectopic calcification is explicable on the basis of a primary magnesium deficiency that increases susceptibility to toxicity of calcemic agents and ectopic calcification.
12.5.2 Hypophosphatasia
The term "hypophosphatasia" has been applied to the inborn error of metabolism that is characterized by defective bone mineralization, associated with low serum alkaline phosphatase activity, and high urinary output of phosphoethanol amine (Reviews: Fraser, 1957; Currarino, 1957). Most of the reports of this condition also indicate hypercalcemic values. No data have been found on magnesium levels, but there is reason to suspect that magnesium deficiency might be contributory to development of this syndrome.
Experimental magnesium deficiency causes low levels of alkaline phosphatase activity in bone, as well as in serum. Rats surviving few to 28 days of magnesium deficiency, and then repleted, had fragile bones thereafter (Duckworth et al., 1940), such as are seen in adults whose hypophosphatasia is diagnosed late (Fraser, 1957). Low levels of bone alkaline phosphatase have been reported in patients with hypophosphatasia (Rathbun, 1948; Sobel et at., 1953; Engfeldt and Zetterstrom, 1954; Schlesinger et al., 1955; Currarino et al., 1957). Without optimal amounts of alkaline phosphatase in bone, its mineralization is inhibited, since alkaline phosphatase is necessary for local destruction of mineralization inhibitors, such as polyphosphates and pyrophosphates. Additionally, high levels of phosphates intensify magnesium deficiency and have been correlated with increased tendency toward bone demineralization, possibly mediated by both mechanisms: (1) lowering of alkaline phosphatase levels caused by magnesium deficiency, and (2) exceeding the capacity of the phosphatase available to destroy the excess phosphates.
Osteopenia, associated with hypophosphatasia, has developed in utero, as well as in infancy, childhood, and adult life (Rathbun, 1948; Sobel et al., 1953; Engfeldt and Zetterstrom, 1954; Schlesinger et al., 1955; Fraser, 1957; Currarino et al., 1957; Beisel et al., 1960; Lessell and Norton, 1964; Pourfar et al., 1972; Rudd et al., 1976). The most severe form is among those whose clinical manifestations develop earliest, possibly beginning in utero. Extensive osteopenic lesions that are found at birth, or in the early months of life, resemble those of osteogenesis imperfecta. Affected infants are assumed to have had spontaneous fractures that healed imperfectly and with angulation (Fraser, 1957). Similar fractures have been reported among infants vulnerable to prenatal and early infantile hypomagnesemia, particularly those born to preeclamptic women and to immature mothers with frequent or multiple pregnancies. Intrauterine growth retardation of abnormal pregnancies and placentas might give rise to fetal hypomagnesemia that can play a role in bone dysplasia. Possibly contributory is vitamin D administration during pregnancy, which has been shown to increase placental scarring in women. Hypervitaminosis D in pregnant rats has been shown to damage the placenta and has been implicated in the bone damage of the pups: thin bones with abnormal osteoid and spontaneous fractures. The lesions, like those of early severe hypophosphatasia, were considered similar to those of osteogenesis imperfecta, and were speculated to have been caused by passage of excessive vitamin D to the fetus through the damaged placenta (Ornoy et al., 1968, 1972). That excessive vitamin D can damage the osteogenic process, leading to lesions very much like those of severe early hypophosphatasia, was shown in 1932 by Shelling and Asher. Young rats on a diet that increased susceptibility to vitamin D toxicity (low in calcium and high in phosphate) showed progressive demineralization and replacement of trabeculae by osteoid remnants and tiny fragments of calcified material when they were given excessive vitamin D for 26 days. It is conceivable that fetuses of pregnant women who are hyperreactive to vitamin D, who consume excessive phosphate-containing foods and beverages, and who are magnesium deficient are at particular risk of developing bone dysplasia.
Possibly the unexplained convulsions of infants with early severe hypophosphatasia (Rathbun, 1948; Fraser et al., 1957; Currarino et al., 1957) might also be of hypomagnesemic derivation, such infants probably having poor skeletal magnesium reserves to meet the requirements of early life (especially in those who are fed cows' milk). The infants commonly suffer from irritability, anorexia, and persistent vomiting, and among those surviving to the second year, craniostenosis develops (Fraser, 1957). These manifestations again focus on the possible role of abnormal response to vitamin D as a contributory factor. They are comparable to those of infantile hypercalcemia, associated with hyperreactivity to vitamin D (Review: Seelig, 1969b), in which low levels of serum alkaline phosphatase have also been reported (Lightwood, 1932; Fanconi and Girardet, 1952; Schlesinger et al., 1956; Amann, 1959; Illig and Prader, 1959; Mitchell, 1960; Editorial, Lancet, 1960; O'Brien et al., 1960; N. David et al., 1962; Garcia et al., 1964; Fraser, 1966). Among 14 patients with hypercalcemia, not of hyperparathyroid origin, N. David et al. (1962) recorded low alkaline phosphatase in five with vitamin D toxicity or hyperreactivity. Another similarity of hypophosphatasia and hypervitaminosis D is the development of band keratopathy (Lessel and Norton, 1964) and chondrocalcinosis (O'Duffy, 1970) in hypophosphatasia and in vitamin D toxicity (J. E. Howard and Meyer, 1948; Chaplin a al., 1951, B. Andersen, 1956). In both hypophosphatasia and infantile hypercalcemia there is greater than normal susceptibility to vitamin D toxicity (Sobel et al., 1953; Reviews: Fraser, 1957; Seelig, 1969b), but the skeletal abnormalities of hypophosphatasia have not responded to vitamin D therapy (Engfeldt and Zetterstrom, 1954; Fraser, 1957). It is speculated that magnesium deficiency might underlie both the susceptibility to vitamin D toxicity, and the vitamin D resistance of the hypophosphatasia syndrome. Magnesium has been protective against development of cardiovascular and renal lesions of vitamin D toxicity. Yet, in magnesium deficiency there is vitamin D resistance.
In view of the fact that vitamin D excess causes magnesium depletion, it is of interest that chondrocalcinosis has also been reported in patients with hypomagnesemia and in experimental magnesium deficiency and phosphate loading, as well as in hypervitaminosis D (Christensen et al., 1951; DeWind, 1961). Vitamin D increases renal tubular reabsorption of phosphorus (Harrison and Harrison, 1941), as well as magnesium loss.
Nephrocalcinosis is common to hypophosphatasia (Review: Fraser, 1957), hypervitaminosis D (Review: Seelig, 1969b), and magnesium deficiency. The most notable difference between hypophosphatasia and infantile hypercalcemia is the osteopenia of the former and the osteosclerosis of the latter. It should be noted that vitamin D toxicity in animals on high intakes of calcium, and in children (most of whose vitamin D is in milk, which is rich both in calcium and phosphate), tend to have hypermineralized bones. Vitamin D and its metabolites, however, have bone mineral-mobilizing activity, and vitamin D toxicity in adults is generally associated with osteomalacia. High phosphate intakes are also implicated in osteopenia.
Several additional similarities to abnormal findings of magnesium deficiency have been reported in hypophosphatasia. The teeth are irregularly calcified and tend to be lost prematurely, a finding attributed to inadequate growth of alveolar bone (Fraser, 1957). Comparable changes have been described in magnesium-deficient rats (Bernick and Hungerford, 1965; Trowbridge and Seltzer, 1967) and hamsters (Yamane, 1962, 1970), and both spontaneously and experimentally in several species of animals when given diets high in phosphates.
Children with hypophosphatasia, whose lesions become apparent after the age of six months, generally have less severe bone lesions. They are characterized by coarse metaphyseal trabeculae that are distorted and irregularly calcified. Bernick and Hungerford (1965) described comparable lesions in magnesium-deficient rats. Possibly the two brothers with epiphyseal irregularities and areas of long bone rarefaction, who had hypercalcemia and hypophosphatasia, and were diagnosed as a rare form of renal rickets because of excessive renal tubular reabsorption of phosphorus (Schneider and Corcoran, 1950), might have had abnormal metabolism of magnesium, vitamin D, or both.
High urinary output of phosphoethanolamine is characteristic of patients with hypophosphatasia (Fraser, 1957). In view of the foregoing correlations of findings of this metabolic disorder, with some of those of magnesium deficiency, it is of interest that magnesium deficiency has caused urinary excretion of phosphoethanolamine in a third of the animals in which this parameter was explored, and that several magnesium-deficient patients also had both low serum alkaline phosphatase levels and high urinary outputs of phosphoethanolamine (Pimstone aet al., 1966). An unpublished observation of high phosphoethanolamine excretion in a woman with latent tetany of magnesium deficiency (Seelig et al., 1975) is of interest. In addition to excreting about 21/2 times more than normal amounts of phosphoethanolamine, she also had low alkaline phosphatase levels following a trial period of 25-OH-D3 therapy, during which her serum magnesium fell further. Her magnesium deficit has not been reparable because she is a renal magnesium waster (Seelig et al., 1976/1980).
12.6.1. Osteoporosis
There have been few studies on the influence of hormonal imbalances on bone magnesium accretion in postmenopausal osteoporosis, the most common cause of this disease. It has been estimated that no fewer than 6,000,000 have this disease in the United States (Harris and Heaney, 1969), even on the basis of the crude measure of osteoporosis provided by roentgenograms (which detects vertebral osteopenia only with loss of 30% to 50% of skeletal mass). The abnormalities in skeletal renewal that occur with metabolic bone disease and hormonal imbalances have been evaluated by Harris and Heaney (1969). Only those facets pertaining to possible mediating effects of magnesium loss in the hormonal imbalances are considered here. The available data suggest that magnesium loss from bone might contribute to several forms of osteoporosis.
Considering the effects of estrogen (and other female sex hormones), a deficiency of which has been most implicated in postmenopausal osteoporosis, and treatment with which has been and is under trial, there are fragmentary data that suggest that its effects on magnesium might be responsible for both promising and adverse effects. A clue to the retention of magnesium caused by estrogen was uncovered when analysis of metabolic studies showed that young women retain more of a marginal magnesium intake than do young men (Seelig, 1964). This observation was confirmed by Amiot et al. (1969) and Durlach (1970) in normal subjects and in patients with osteopenias. Comparable studies of magnesium retention of postmenopausal women have not been found. It was postulated that this difference in retention of magnesium might be a factor in the greater resistance of young women than men to cardiovascular disease (Seelig, 1964; Seelig and Lehr, 1971/ 1973; Seelig and Heggtveit, 1974), and might contribute to the increase in incidence of both cardiovascular and bone disease after the menopause (Seelig and Lehr, 1971/1973). On the other hand, plasma magnesium levels tend to be higher in young men than in young women, particularly during the period of greatest estrogen secretion, or when they are taking oral contraceptives (Dahl, 1950; N. Goldsmith, 1963; N. Goldsmith and Goldsmith, 1966; N. Goldsmith and Baumberger, 1967; DeJorge et al., 1967; Durlach, 1970; N. Goldsmith et al., 1970; Olatunbosun et al., 1974; 1976/1978; N. Goldsmith and Johnston, 1976/1980). Durlach (1970) cautioned that this effect of estrogens might contribute to thromboembolic phenomena, and recommended that women on oral contraceptives be given magnesium concomitantly to prevent increased coagulability that might be caused by lowered plasma magnesium levels. N. Goldsmith and Johnston (1976/1980) have reviewed the evidence as to the risk of thromboembolism in women on oral contraceptives.
Estrogen exerts both direct and indirect effects on bone metabolism. It inhibits bone resorption in vitro(P. Stern, 1969) and increases endosteal bone formation in mice (M. Silverberg and Silverberg, 1941), but decreases calcium accretion in rats, even though it decreases bone resorption (Lindquist et al., 1960). Estrogens antagonize PTH-induced bone resorption (Ranney, 1959; Nordin et al., 1970; Atkins et al., 1972), and in ovariectomized rats the bone-resorptive effect of PTH is increased (Orimoet al., 1972). Since PTH mobilizes bone mineral (including magnesium), estrogen has increased bone uptake of magnesium (N. Goldsmith and Baumberger, 1967), and magnesium deficiency causes osteopenia, it is possible that at least part of the effect of estrogen on bone might be mediated by its effect on bone magnesium levels. Another bit of evidence that implicates magnesium loss in some of the osteopenic processes is the degranulation of mast cells in magnesium deficiency (Hungerford and Karson, 1960; Bois, 1963), a process that causes release of heparin as well as histamine. Heparin enhances the resorptive response of bone to PTH (Goldhaber, 1965). Increased bone sensitivity to PTH has been implicated in osteoporosis, even in the absence of elevated endogenous PTH levels (Heaney, 1965; Harris and Heaney, 1969). Further support for this concept has been provided by Bélanger et al. (1975), who confirmed the damage to mast cells caused by magnesium deficiency, showed that female rats are more susceptible to magnesium deficiency-induced mast cell damage than are males, and that estradiol in the females and testosterone in the males resulted in less mast cell depletion.
Thus, there are data, deriving from magnesium-deficiency studies, that bear on some of the mechanisms that might be involved in the clinical benefit that has been reported with long-term prophylactic use of estrogens in postmenopausal women. Henneman and Wallach (1957) reviewed the records of 200 patients given estrogens by Albright and his colleagues for 1 to 20 years and found that, using loss of height as an index of osteoporosis, the use of estrogen arrested further loss of height in those already suffering from the disease, and prevented height loss in those whose postmenopausal estrogen treatment had begun before there was evidence of osteoporosis. [In regard to the concern about estrogen increase of cancer, the authors commented that in this group of 200 patients, who were given intermittent (cyclic) therapy, the incidence of carcinoma of the breast, cervix, and endometrium was low.] Determination of the effect of estrogens on bone thickness by means of densitometry has also shown estrogens to inhibit progression of postmenopausal osteoporosis (Meema and Meema, 1968; M. E. Davis et al., 1966; Meema et al., 1975; N. F. Goldsmith and Johnston, 1975,1976/1980). Estrogen has also been shown to decrease bone resorption, as measured by urinary output of hydroxyproline (Riggs et al., 1969; Gallagher and Nordin, 1972) and to be effective (in doses of no less than 1.25 mg of conjugated estrogen in a series of 220 severely osteoporotic women) in arresting vertebral fractures (Gordan, 1971). The mechanism of action has been postulated to be via estrogen inhibition of PTH-induced bone resorption in post- menopausal women (Nordin, 1971; Gallagher and Nordin, 1972; Seelig and Lehr, 1971/1973).
Why women are more susceptible than are men, in the middle years, to (presumed) relative hyperparathyroidism is not clear. It is possible that the estrogen-induced lowering of plasma magnesium (which might be the result of a shift to intracellular sites) might result in chronic stimulation of parathyroid secretion. If such stimulation causes parathyroid hyperplasia [as Larvor et al. (1964a) have demonstrated in calves], when the ovaries cease functioning the overactive parathyroids might continue to mobilize bone minerals, excessively in the absence of the counteracting effect of estrogen (Fig. 12-6).
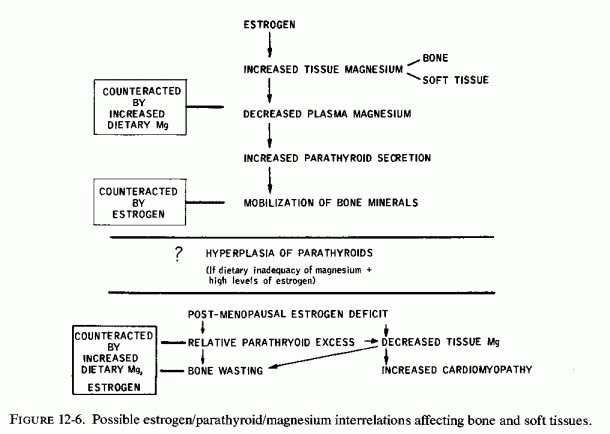{kind=link}
The later development of osteoporosis in men probably reflects their longer gonadal activity. Testosterone has also been shown to have activity in clinical osteoporosis (Gordan, 1954).
Evidence that calcitonin (CT) retards disuse osteoporosis (Hantman et al., 1973) and that magnesium administration stimulates CT secretion suggests that magnesium administration may be useful in this form of osteoporosis. It recalls the work with rats, showing interrelationships among magnesium, CT, PTH, and cortisone (Palmieri et al., 1969; Eliel et al., 1971). Cortisone, an excess of which has long been known to cause osteoporosis, abolished the hypomagnesemic effect of CT, an effect attributed to its interference with CT inhibition of bone-resorption. On the other hand, patients with regional enteritis had their magnesium deficiency (to the point of hypomagnesemia) intensified by corticosteroid therapy (Gerlach et al., 1970). Although only the acute signs of magnesium deficiency were considered in that paper, it should be recalled that malabsorption is implicated in osteopenia (i.e., celiac rickets and osteomalacia), as are corticosteroid therapy and magnesium depletion.
Administration of magnesium supplements to several patients, including a few with conditions (e.g., alcoholism or cirrhosis) that predispose to magnesium deficiency, improved their calcium retention (Briscoe and Ragan, 1966). Du Ruisseau and Marineau (1971/1973) showed that patients with osteopenia retained more calcium when supplemented with magnesium. In contrast, administration of calcium to such patients increased their magnesium deficit (Table 12-4, Fig. 12-7, Parlier et al.,. 1963; Amiot et al., 1969).
{kind=link}
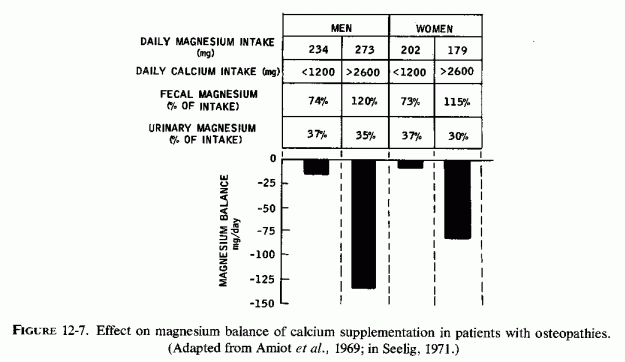{kind=link}
12.6.2. Renal Osteodystrophy
The osteopenia seen in patients with uremia, whether they are dialyzed or not, entails complex interrelationships among etiologic and complicating factors; space does not permit their consideration here. Selected from the massive literature on this subject are data directly or indirectly bearing on the possibility that tissue magnesium depletion might play a role. Serum magnesium levels are unreliable as an index of the magnesium status of uremic patients. High normal, and low levels have been reported that are unrelated to tissue levels (Lim et al., 1969a; Lim and Jacob, 1972c). Metabolic balance determinations are also unreliable, the equilibrium reported by Clarkson et al. (1965) being associated with subnormal intestinal magnesium absorption that balanced its subnormal urinary excretion. In contrast, patients with chronic renal failure receiving low protein diets, lost as much as 139 mg of magnesium daily, despite slightly elevated serum magnesium levels (Kopple and Coburn, 1973).
Massry and Coburn (1970a) proposed that tissue magnesium deficiency in patients with progressive renal failure (even when serum magnesium levels are elevated) might be contributory to their hypocalcemia, vitamin D resistance, and defective response of the skeleton to parathyroid hormone. On the other hand, if the tissue magnesium deficit involves the parathyroids, secondary hyperparathyroidism might ensue. Pletka et al. (1971) have, in fact, shown that the levels of parathyroid hormone (PTH) of patients treated by hemodialysis with water containing 1.5 to 2.5 mEq of magnesium per liter fell 20% from pretreatment high levels. Those who were hemodialyzed with water containing low concentrations of magnesium (0.5 mEq/liter), who had lesser initial elevations of PTH, showed a 118% rise in PTH after two months of treatment. It is well known that patients being treated by dialysis are subject to hyperparathyroidism, with the attendant problems of bone loss and metastatic calcification (Buckle, 1970; Kleeman et al., 1970; Genuth et al., 1970; Danesh et al., 1970; Terman et al., 1971; Henderson et al., 1971; Editorial, Brit. Med. J., 1972; Arora et al., 1975). Cardiovascular involvement is common, and cardiovascular disease is by far the leading cause of death in patients on chronic dialysis (Lowrie et al., 1974), accelerated arteriosclerosis (Lindner et al., 1974; Curry and Roberts, 1977), myocardial calcification, and heart block having been reported.
There is concern about producing hypercalcemia and possibly hypermagnesemia by using untreated hard water (Freeman et al., 1967). Acute symptoms of hypermagnesemia (blurred vision, flushed face, weakness, and inability to stand) were produced by use of a dialysate containing 15 mEq of magnesium per liter (Govan et al., 1968). Posen and Kaye (1967) have reported that magnesium levels in the dialysis bath water in major centers range from almost 0 to 0.8 mEq/liter, usually depending on the concentration in the water supply; although they used Montreal water (one of the harder water supplies available), they added 1 mEq of magnesium to each liter, so as to provide 1.65 mEq/liter. They attribute to the added magnesium the freedom of their patients from metastatic calcification over the four-year observation period. They did not comment on the incidence of osteodystrophy, but Catto et al. (1973) commented that osteodystrophy is not a problem in Montreal or London (both hard-water cities), whereas it is in Newcastle and Los Angeles. Kleeman et al.(1970), who commented on the magnesium supplied by two medical centers in Los Angeles, 0.5 and 1.5 mEq/liter, suggested that providing a dialysate magnesium concentration (1.5 mEq/liter) sufficient to prevent hypomagnesemia might reduce the tendency toward metastatic calcification and secondary hyperparathyroidism. This recommendation was also made by Danesh et al. (1970). It may be relevant that the accelerated arteriosclerosis reported by Lindner et al. (1974) was from a center in Seattle, a soft-water area. The low incidence of osteodystrophy in two hard-water cities (Catto et al., 1973) suggests that the magnesium provided might also protect against secondary hyperparathyroidism and osteodystrophy.
However, there is no consensus as to the optimal magnesium concentration of water used for dialysis. Unlike Posen and Kaye (1967), who added magnesium to the hard water, it is common to attempt to bring the serum magnesium levels down to normal limits by using dialysates with low magnesium concentrations (from 0.16 to 1.0 mEq/liter) (Johny et al., 1971; W. K. Stewart and Fleming, 1971; 1973; Paschen et al., 1971). In one reported patient receiving twice-weekly hemodialysis with water containing 0.8 mEq/liter of magnesium, severe hypomagnesemic cramps developed that promptly responded to magnesium therapy (Triger and Joekes, 1969). In view of the cited evidence that tissue levels of magnesium can be low in patients with renal disease, despite high serum levels, and the importance of tissue magnesium in protecting against pathologic changes in cardiovascular and skeletal tissues, the advisability of depleting the body magnesium by use of low magnesium dialysate is open to question.
Since bone and serum magnesium tend to be in equilibrium, the fact that surface bone magnesium levels of uremic patients tend to be higher than normal (Contiguglia et al., 1972; A and Miller, 1973) is not surprising. Its significance is uncertain. Alfrey and Miller (1973) found that 30% of the bone magnesium of uremic patients with hypermagnesemia is within the bone hydration shell or on the crystal surface, and speculate that, since magnesium can influence crystal size and stability, an excess might play a role in osteodystrophy. They noted, however, that the deeper magnesium is not as readily exchanged, and its mobilization is dependent on the resorptive process. However, chronic experimental uremia adversely influences collagen metabolism in both skin and bone (Hahn and Avioli, 1970). Also, experimental magnesium depletion causes formation of abnormal bone matrix with defective calcification capacity. Thus, it seems likely that loss of deep-located bone magnesium should have a more significant effect on the osteopenia of renal disease than the gain at the surface.
A final indirect bit of evidence that magnesium deficiency might contribute to renal dystrophy is the observation that renal osteodystrophy is rare in Israel (Berlyne et al., 1973b). The rarity of this disease (in Beer Sheva) was attributed by the investigators to the low phosphorus intake of dwellers in that area. However, in another publication, Berlyne et al. (1973a) reported that the water in that area was also very high in magnesium and calcium.
12.7.1. Osteochondrosis
There are meager clinical data that suggest that magnesium deficiency might play a role in osteochondrosis or osteochondritis (Legg-Perthes disease; slipped epiphyses). J. F. Miller (1944) reported a child who had had neonatal tetany and hyperirritability and cyanotic episodes during the early weeks of life, for which he was given calcium therapy, which was continued (with halibut liver oil) from then on. By 6 months of life he developed normocalcemic convulsions that stopped at the age of one; his tremors persisted. At 31/2 years of age he had osteochondrosis of the capital epiphysis of the left femur, including fragmentation and flattening of the epiphysis. At that time he had hypercalcemia (12.9 mg/100 ml). By the age of 6 years, in addition to dizziness and tremors, he had developed muscle cramps and carpopedal spasms, at which time his plasma magnesium was determined for the first time; it was 1.4 mEq/liter. He responded strikingly to magnesium therapy (300 mg MgSO4 three times daily), with disappearance of tremors and dizziness. After the supplements were stopped by his parents for a week when he suffered an attack of bacillary dysentery, his tremors and dizziness recurred, he had positive Trousseau and Chvostek signs, and his plasma magnesium dropped to 0.5 mEq/liter. He again improved promptly on magnesium therapy. The osteochondrosis had been treated surgically, and thus the effect of the magnesium therapy on this disease could not be ascertained, but Miller speculated that there might have been a relationship between the boy's probable early magnesium deficiency and his epiphyseal abnormality. Klingberg (1970) reported mild osteochondritis of shoulders, knees, and hips (Legg-Perthes-like) in a boy who suddenly developed a carpopedal spasm of 6 hours duration at 5 years of age and who was found to have both hypomagnesemia (0.8 mEq/liter) and hypokalemia (2.8 mEq/liter). A 6-day metabolic balance study showed minimal negative magnesium balance; supplementation with 60 mEq magnesium (as the acetate) produced a slightly positive magnesium balance (+42 mEq) he continued to excrete 5-9 mEq/day in his urine. After 6 months of magnesium supplementation, the patient's bony lesions reverted to almost normal. With the lower magnesium supplements, his tetany recurred. The possibility of a renal tubular defect in magnesium reabsorption was proposed as an explanation of the child's high magnesium requirement. Follow-up of this child for 6 years has shown persistence of his renal wastage of magnesium. He has also developed cardiac and skeletal abnormalities (W. O. Klingberg, personal communication, 1978). Children with hypophosphatasia (proposed as related to magnesium deficiency) also have metaphyseal and epiphyseal abnormalities, as have some children with vitamin-D refractory rickets, also related to magnesium deficiency.
The studies that show abnormalities of metaphyseal trabeculae and of the epiphyseal cartilage and ground substance (Yamane, 1962; 1970; Yamane and Singer, 1953; Bernick and Hungerford, 1965; Clark and Bélanger, 1967; Trowbridge and Seltzer, 1967; Trowbridge, 1971) provide direct evidence that experimental magnesium deficiency causes abnormalities in epiphyseal structure. Abnormal epiphyseal cartilage and diaphyses have also been seen in pups of pregnant rats overdosed with vitamin D (Ornoy et al., 1972), and cessation of epiphyseal osteogenesis in young rats with vitamin D toxicity (Shelling and Asher, 1932; Ham and Lewis, 1934; Storey, 1960), lesions that might reflect secondary magnesium deficiency.
Before a generalization can be drawn (correlating clinical epiphyseal disease with early magnesium deficiency), there should be evaluation of children with this disease, and of their families, for abnormalities in magnesium absorption and retention.
Enlargement of the joints and marked stiffness were identified as signs of magnesium depletion in calves and were shown to resolve when magnesium salts were added to a high-phosphate, low-calcium diet by Huffman et al. (1930). Deletion of the magnesium carbonate supplement resulted in recurrence of the stiffness within two months. Cattle foraging in low-magnesium areas also developed articular damage, in these instances characterized by erosions of the cartilage (Willers et al., 1965). House and Hogan (1955) demonstrated that optimal intakes of magnesium (0.35% of diet) and potassium (1.5% of diet) prevented the stiffness and periarticular deposition of calcium phosphate that developed in magnesium-deficient guinea pigs receiving only slightly more phosphorus than calcium (P/Ca = 0.9/0.8%) (Hogan et al, 1950). Joint stiffness was worst in guinea pigs fed rations containing 1.7% phosphorus, 0.9% calcium, 0.04% magnesium, and 0.41% potassium.
Chondrocalcinosis has also been associated with human diseases associated with magnesium loss. The first instances were in rheumatoid arthritis patients taking excessive amounts of vitamin D (Review: Christensen et al., 1951). Additional to the metastatic calcification of the arteries, kidneys, and other viscera, there was sometimes marked and disabling calcification of the periarticular structures, involving the synovial cavities, bursae, and tendon sheaths (accompanying generalized osteoporosis). Withdrawal of the toxic vitamin D supplements resulted in decreased periarticular calcification. This condition was usually seen among rheumatoid arthritis patients who had self-medicated themselves with vitamin D supplements providing as much as 200,000 units daily. It was also seen in a child who had been given high dosage (> 40,000 units/day) vitamin D since the age of three because of suspected rickets, diagnosed on the basis of a "peculiar feeling to the skull," as well as wrist changes. When seen by the investigator (DeWind, 1961) at 5 he had periarticular calcification, as well as osteosclerosis that encroached on the medullary canals. The bone changes resemble those described in experimental magnesium deficiency.
A patient with monoarticular osteoarthritis, whose hypophosphatasia was diagnosed in middle age, had calcification of the articular cartilage of her hips, symphysis, and arthritic knee (O'Duffy, 1970). This was the first time note was taken of the deposition of calcium pyrophosphate in cartilage of a patient with hypophosphatasia, but O'Duffy reviewed the literature and found several additional cases in which periarticular calcification was noted in the case reports. He reviewed some of the metabolic disorders in which pseudogout was reported, and found that it was common in hyperparathyroidism. McCarty et al. (1974), who compared the frequency of concomitant chronic diseases in patients with pseudogout and in those with osteoarthritis of the large weight-bearing joints, found no significant differences, and that immunoreactive parathyroid hormone was elevated in both groups. They postulate that sustained low-grade hyperparathyroidism might be related to the genesis of the articular lesions. This is a provocative observation, since both vitamin D excess and hyperparathyroidism are associated with loss of magnesium. McCarty (1974) and his co-workers (McCarty et al., 1971) have related magnesium with inhibition of calcium pyrophosphate precipitation in synovial fluid, correlating this effect with magnesium-activation of pyrophosphate transphosphorylase.
Precipitation of calcium pyrophosphate in the joints of patients with hypomagnesemia has been reported. McCarty et al. (1974) reported one such instance in a patient with psoriasis. Runeberg et al.(1975) reported a young man who had renal tubular magnesium wasting, hypomagnesemia, and from whose knee joint calcium pyrophosphate crystals were obtained. This patient is of particular interest, since he had had nephrocalcinosis from the age of seven, following calcium therapy of his convulsive hypocalcemia, and developed ECG changes similar to those seen with magnesium depletion when he was 14 years of age. After he retained 247 mmol of magnesium (during a period of intravenous infusions of 4 mmol of magnesium chloride daily for 8 days), he was maintained on high oral magnesium dosage (20mmol as Mg (OH)2 and 30 mmol as MgCl2 and potassium for 2 years. His joint effusion disappeared and he remained symptom-free since. Rapado and Castrillo (1976/1980b) reported a man of 38 with knee joint pain and swelling of several years duration, who also had renal tubular magnesium wasting and hypomagnesemia. He had X-ray evidence of linear calcification of the cartilage, and biochemical demonstration of calcium pyrophosphate in a synovial biopsy. This patient, too, responded to magnesium therapy, but his response is not as clear-cut because he was maintained also on antiinflammatory drug therapy
Ankylosing hyperostosis, a common disorder of the middle-aged and elderly that affects the spine and large joints, has also caused calcaneal spurs (particularly of the heel), and has also been associated with precipitation of crystals of calcium pyrophosphate dihydrate. Among 30 patients reported by Utsinger et al. (1976), one had hypomagnesemia, three had hyperphosphatemia, and four had elevated serum alkaline phosphatase. More intensive study of the magnesium status should yield useful data.
Complicating the problem of chondrocalcinosis and exostosis and their response to magnesium is pyrophosphate's inhibition of precipitation of calcium phosphate compounds in urine (Fleisch and Neuman, 1961; Fleisch and Bisaz, l962a), and the necessity of pyrophosphatase for normal (bone) mineralization. Thus, there must be a delicate balance between enzymes and substrate on the one hand, and concentrations of the minerals: magnesium, calcium, and phosphorus on the other. Hydroxyapatite crystals are most commonly found in periarticular disease, in contrast to the calcium pyrophosphate dihydrate that is more frequent in intraarticular disease, such as is not uncommon in uremia (Parfitt, 1969). Mirahmadi et al. (1973) reported that calcium hydroxyapatite has precipitated periarticularly in renal failure patients undergoing hemodialysis: They suspect that hyperphosphatemia is the most likely provocative factor. They noted that none of their seven patients with this complication had magnesium depletion, and recommended measures to lower the serum phosphate levels and use of higher calcium concentrations in the dialysate, to suppress parathyroid secretion. Since increased magnesium also suppresses the parathyroid function, further study and individualization of the prophylactic or therapeutic regimen is advisable.
Ankylosing spondylitis, accompanying bone resorption (from adolescence on), and irregularity and erosion of the articular cartilage (such as has been reported in magnesium deficiency, supra vide), with obliteration of the joint space, has been encountered in primary hyperparathyroidism (Bunch and Hunder, 1973), a condition associated with magnesium loss.
Articular lesions-peri-, para-, and intraarticular calcification-have also been seen in uremic patients (Review: Parfitt, 1969). An unusual paraarticular lesion occasionally seen in such patients, and that had been more common when high doses of vitamin D were given as treatment for arthritis (Christensen et al., 1951) is tumoral calcinosis, rubbery or cystic calcific mass. McPhaul and Engel (1961) reported two patients with this disorder in one family, four of whom-including the patients-had low plasma alkaline phosphatase levels. Parfitt (1969), who reviewed the factors involved in soft tissue calcinosis of uremia (including the articular forms calcification), considers hyperphosphatemia the most important single factor, and calciphylaxis (produced by prior "sensitization" with vitamin D or parathyroid hormone) as a unifying hypothesis. Since Selye, who promulgated the calciphylaxis theory, found magnesium to be protective (against cardiorenal calcinosis caused by high phosphate, vitamin D, or PTH), perhaps low magnesium levels caused by these agents (as well as by uremic acidosis) might play a role in the abnormal calcification processes of the joints in uremic patients. Whether magnesium deficiency also plays a role in osteoarthritis cannot be averred in the absence of other than the meager animal and clinical data available.
Leonard and Scullin (1969) and Leonard et al. (1971) have proposed that in soft tissue, where the concentration of magnesium exceeds that of calcium, the formation of MgATP inhibits calcium apatite formation. This group has demonstrated that the local magnesium/calcium ratio influences calcification of tendons (in turkeys) and that egress of magnesium precedes the onset of calcification (Leonard et al., 1976). This is a physiologic maturation process in turkey tendon. It seems plausib1e that low Mg/Ca concentrations in soft tissues, and in articular and periarticular tissues, might similarly participate in calcification, and that a higher magnesium concentration might inhibit it.
Damage to teeth, as well as to bones and to soft tissues, were among the findings reported from the earliest magnesium deficiency studies. Kruse et al. (1932) found that rats surviving severe magnesium deficiency for 10 weeks had loose molars and incisors. Further data on the abnormal periodontal soft tissues were provided by H. Klein et al. (1935). Brittle, chalky teeth (loose in their sockets) were noted by Watchorn and McCance (1937) in subacute magnesium-deficient rats. They, like H. Kleinet al. (1935), found striations in the dentin, suggestive of intermittent interference with the calcification process; they also reported odontoblastic degeneration. Becks and Furata (1939, 1941) reported pronounced degenerative changes in the enamel epithelium of rats by the 72nd day of subacute magnesium deficiency. Irving (1940) confirmed the damage to the enamel, caused by magnesium deficiency, as well as striations in the dentin. He also noted increased width of the predentin above the basal portion of the teeth that he considered unique to magnesium deficiency. Yamane and Singer (1953) found alternate bands in the incisors of magnesium-deficient hamsters that were associated with odontoblastic degenerative changes, and decreased width of the enamel-forming cells (Yamane, 1962). Bernick and Hungerford (1965) showed that magnesium-deficient rats had disturbed dentin calcification. It was characterized by a wide uncalcified zone separated from the predentin by a thin calcified line. There was also odontoblastic degeneration. Trowbridge (1971) and Trowbridge et al. (1971) point out that magnesium deficiency also causes dentinal striations and that the incisal dentinal striations ceased within four days of magnesium supplementation; thereafter the new dentin was normal except in areas adjacent to enamel, where it was somewhat attenuated.
The importance of magnesium for the metabolism of teeth is suggested by the avidity of teeth of control magnesium-deficient lambs for 28Mg (McAleese et al., 1961). F. R. Morris and O'Dell (1961) had shown that increasing the phosphorus content of the diet from 0.4 to 1.7% did not affect the calcium or phosphorus content of the teeth but intensified magnesium deficiency, which they had earlier shown to cause formation of the defective teeth and decay; the teeth were loose in their sockets (O'Dell et al., 1960). The authors commented that their findings suggested that the magnesium deficiency probably affected cell function and development of the organic matrix of the tooth, rather than its mineralization. That the organic matrix of dentin of magnesium-deficient rats did, indeed, differ from that of controls was demonstrated by Bernick and Hungerford (1965). Differences in staining characteristics suggested that the ground substance of the matrix of bones and teeth of magnesium-deficient rats contained less polymerized mucopolysaccharides; they are thus less subject to normal calcification. Defective dentin matrix formation by acutely magnesium-deficient rats was confirmed by Trowbridge and Seltzer (1967). They demonstrated greatly reduced tritiated proline labeling in the organic matrix of the dentin and retarded dentin formation and calcification, arrested appositional bone growth and resorption of the crest of the alveolar process (Trowbridge and Jenks, 1968; Trowbridge, 1971). The periodontal ligament was wider in the magnesium-deficient rats than in the controls, and there was minimal osteoblastic activity and lesser evidence of alkaline phosphatase activity in the pulps and the serum of the deficient rats. Magnesium-deficient hamsters also had periodontal abnormalities, as compared with pair-fed controls (Yamane, 1962, 1970). The periodontal ligament was disorganized, calculi formed in the gingival sulci, and the interdental bony septum showed resorption. Following extraction of teeth, the magnesium-deficient hamsters exhibited delayed healing, an effect attributed to impaired matrix formation. Delayed eruption of the permanent teeth, as well as abnormal mineralization of both dentin and enamel, odontoblastic degeneration, arteriosclerosis of pulpal vessels, and pulpal calcification were reported by Binus (1968) in magnesium- deficient dogs.
Two genetic clinical abnormalities that the author postulates may be associated with magnesium depletion: hypophosphatasia (Reviews: Fraser, 1957; Currarino et al., 1957) and pseudohypoparathyroidism (Review: Bronsky et al., 1958) are associated with dental disorders that bear some resemblance to those of experimental magnesium deficiency. In hypophosphatasia irregular calcification and severe caries have been reported. Three quarters of the children whose disease became manifest by the sixth month of life had premature loss of teeth, attributed to inadequate growth of alveolar bone and incomplete formation and early resorption of the roots of the teeth (Review: Fraser, 1957; Pourfar et al., 1972). Beisel et al. (1960) reported early loss of all permanent teeth of a patient who presented his first signs of hypophosphatasia as an adult. Both in pseudohypoparathyroidism and in idiopathic hypoparathyroidism, comparable dental abnormalities are not uncommon (Table 12-5) (Bronsky et al., 1958).
It is noteworthy that in two conditions with abnormal vitamin D metabolism, dental abnormalities have been reported. Children with hyperreactivity to vitamin D, who also have hypophosphatasia, have a high incidence of malocclusion, enamel hypoplasia, and severe caries. Rampant caries, necessitating early extraction of all teeth (before the age of 20) has been reported in a patient with familial hypophosphatemic vitamin-D-resistant rickets (Blackard et al. 1962). Enamel hypoplasia involving teeth that calcify after birth was found in members of a family with hypophosphatemic rickets and secondary hyperparathyroidism (Arnaud et al., 1970). On the other hand, periodontal disease has been correlated with high phosphate intakes and secondary hyperparathyroidism and with osteoporosis (Lutwak, 1974; Review: Krook et al., 1975).
-----------------------------
Part III: Chapter 13
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
Metastic calcification, frequently involving the kidneys, is not infrequent in patients with hypercalcemia, whether of dietary or metabolic derivation, because of osteolytic processes, or as a result of therapy. The study by B. S. W. Smith and Nisbet (1968), which showed that magnesium-deficient rats develop nephrocalcinosis, and later osteoporosis, is an appropriate reference for the transition from bone damage to renal damage of magnesium deficiency.
The diets contrived to be magnesium deficient are almost always imbalanced in other constituents as well. The early diets were usually rich in fats, calcium, phosphorus, and vitamin D, which were effective in producing acute signs of magnesium depletion rapidly (Kruse et al., 1932) and also produced severe renal glomerular and tubular damage that was most extensive at the junction of the cortex and medulla (Cramer, 1932; Brookfield, 1934). Modifications of that diet (designed specifically to produce hypercholesterolemia and atherosclerosis) also produced renal damage (Hellerstein et al., 1957; Gottlieb et al., 1959; Vitale et al., 1959). There was deposition of calcium microliths in the lumina of the collecting tubules that was accompanied by tubular dilatation, and flattened epithelium. High dietary magnesium (96mg Mg/100 g of diet) abolished the renal tubular calcification, regardless of the amount of calcium fed, in the animals not loaded with cholesterol and cholic acid, and decreased it in fat-loaded rats.
With less imbalanced diets, designed to produce subacute magnesium deficiency (Watchorn and McCance, 1937), rats developed occasional to more frequent calcareous deposits scattered throughout the renal cortex and medulla. Those with most severe damage had extensive calcareous casts and obliteration of the epithelium of the straight and collecting tubules, but no glomerular changes. Greenberg et al. (1938), also using a less imbalanced diet that did not produce signs of acute deficiency and that contained neither excess phosphate nor very high doses of vitamin D, but was high in calcium (Tufts and Greenberg, 1937), found that prolonged magnesium deprivation of rats produced corticomedullary necrosis and calcinosis involving both the tubular cells and lumina. They attributed the renal calcinosis to the high calcium/magnesium ratio. Greenberg (1939) later attributed part of the severe manifestations of the magnesium-deficiency syndrome (including the renal calcinosis) in the early studies to the inadequacy of vitamins B2 and B6 in the vitamin-B-complex supplements then available. The concomitant magnesium and pyridoxine deficiencies might be relevant to calcium oxalate deposition in the kidneys, magnesium being a cofactor in vitamin B6metabolism (Review: Durlach, 1969b), oxalate excretion increasing in vitamin B6 deficiency (Gershoff et al., 1959), and a combination of high magnesium and vitamin B6 being useful in decreasing calcium oxalate and apatite nephrocalcinosis and urolithiasis (Gershoff and Andrus, 1961; Gershoff and Prien, 1967). Gershoff and Andrus (1961) also showed that the amount of magnesium usually provided control rats (400 ppm) did not completely prevent formation of apatite salts in the kidneys. Tenfold higher intakes were completely protective.
Most of the magnesium-deficiency data derived from rat studies have been obtained with diets rich in calcium and phosphorus, although the marked imbalances in dietary Ca/Mg and P/Mg are rarely noted. Usually they provided from 600/1 to 60/1 ratios of Ca/Mg. For example, rats reported by Hesset al. (1959) were fed a diet delivering 18 mmol Mg/kg of diet and 150 mmol Ca; the deficient group were given 0.25 mmol Mg. They had mitochondrial swelling of tubular cells (observed as early as 3 days of magnesium deprivation) in the distal segment of the convoluted tubule and extending to the thick descending limb. By 6 days, Henle's loop was also involved. Tubular necrosis was noted by 12 to 20 days, and there were calcium deposits intracellularly and in the lumina, forming calcareous casts. The semisynthetic magnesium-deficient diet provided by Mishra (l960a,b) provided a similar Ca/Mg ratio, and caused decreased renal mitochondrial count and increased tubular calcinosis. With an approximately tenfold less disparity between dietary calcium and magnesium, tubular lesions developed in the renal cortex and at the corticomedullary junction by the day 8 of magnesium deficiency (Kashiwa, 1961). Some of the tubular cells were hypertrophied and had vacuolated cytoplasm, others were flattened, and there were numerous calcareous deposits, especially at the corticomedullary junction. Comparable changes, with clumping of renal tubular mitochondria, were correlated with functional renal defects after as little as a week of magnesium depletion (W. O. Smithet al., 1962). The rats exhibited a decreased ability to concentrate and acidify urine and a marked phosphaturia.
Sauberlich and Baumann (1949) found that mice fed diets deficient in thiamine, pyridoxine, or magnesium had aminoaciduria. In a study of chicks and rats (with a Ca/Mg ratio, even in the magnesium-deficient group of rats that was less imbalanced, about 40/1; Bunce et al. (1963) showed that sixfold higher intakes of magnesium were necessary to prevent nephrocalcinosis and aminoaciduria that were seen in the deficient groups. Progressively increased aminoaciduria was also produced in rats on the usual high Ca/Mg dietary ratios of magnesium deficiency studies as the depletion developed (Mazzocco et al., 1966).
Noted in most of the cited magnesium-deficiency studies were the intraluminal calcareous deposits in the corticomedullary area, and the damage to the tubular epithelium. The characteristic early lesion has been described as microliths in the thin limb, the bend of the loop, and the ascending limb of the loop of Henle (ALLH) (Whang et al., 1962; Welt, 1964; Oliver et al., 1966; Schneeberger and Morrison, 1965, 1967; Whang et al., 1969). Ko et al. (t962) reported that rats on a magnesium-deficient diet that provided twice as much calcium as phosphorus developed the typical intraluminal and cellular deposits of calcium phosphate, but that the ALLH was not involved unless there was phosphate loading, as well. Schneeberger and Morrison (1967) showed that the ALLH lesions of magnesium deficiency were intensified by phosphate loads. Similar intraluminal lesions have also been seen in the bend of the loop and in the ALLH of early experimental hyperparathyroidism (Epstein, 1960) and vitamin D toxicity (Epstein et al., 1958; Kent et al., 1958; Veltman, 1959; Potvliege, 1962). This observation is not surprising since both hyperparathyroidism and hypervitaminosis D increase blood and thus urinary loads of calcium, and cause magnesium loss.
Damage to the ALLH by primary or secondary magnesium deficiency creates a situation that intensifies the magnesium deficit. Micropuncture studies have shown that most active renal tubular reabsorption of magnesium occurs at this site (Wen et al., 1970, 1971; Brunette et al., 1974, 1975; Dirks and Quamme, 1978; Quamme et al., 1976/1980). Thus, damage to the cells of the ALLH can cause renal tubular magnesium wasting. The clinical significance of treatment of hypomagnesemic hypocalcemia with calcemic agents or phosphates is discussed elsewhere in this volume.
13.2. Intensification of Magnesium Deficiency and Renal Damage by Excess Vitamin D (Animal)
Vitamin D toxicity, with or without high calcium intakes, has long been known to cause soft tissue damage. The cardiovascular lesions have attracted most notice. Even brain damage and calcification have been described, both in test animals and in infantile hypercalcemia (Review: Seelig, 1969b). How much of the total renal damage of most experimental magnesium-deficiency studies is caused by relative or absolute vitamin D excess, and how much might be due to excess phosphate intake or tubular reabsorption, each of which intensifies magnesium loss and increases mobilization of bone constituents has not been resolved (Fig. 13-1). The answer must await definitive studies that evaluate the effects of each agent, with the others kept at the amounts necessary to avoid inducing specific deficiencies or imbalances. Such might evoke hormonal responses that could obfuscate the effect of the mineral under investigation.
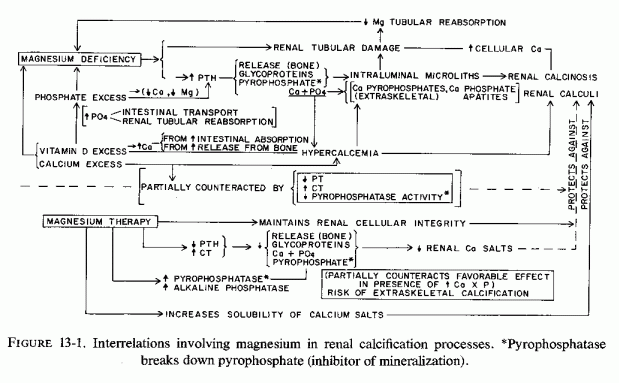{kind=link}
Konetzki et al. (1962) showed sequential accumulation of calcium and mucopolysaccharides in nephrocalcinosis due to vitamin D toxicity. The renal deposition of calcium started before the kidneys began to accumulate radioactively tagged sulfur. After the process had started the 35S uptake intensified. (Giacomelli et al. (1964) observed that calcium deposited as hydroxyapatite crystals intraluminally in the proximal convoluted renal tubules and in the cytoplasmic vacuoles of the tubular cells of rats poisoned by vitamin D. They consider the crystallization process to be induced by deposition of mucopolysaccharides (derived, like the calcium and phosphate, from the bone, dissolution of which is caused by hypervitaminosis D). These changes are very much like those described in magnesium-deficient rats: tubular calcium phosphate on a glycoprotein matrix (Bunce and Bloomer, 1972). As with that of magnesium deficiency, the initial lesion of vitamin D nephrotoxicity is proposed to be cytochemical and cytologic alterations (Scarpelli, 1966). The earliest consistent changes, by light microscopy, was increased atypical cytoplasmic vacuoles in the proximal tubular cells [manifest within 24 hours after a single massive oral dose (45,000 units) of vitamin D]. Slight mitochondria damage was also seen. Intracellular edema and marked cellular distortion developed after four doses. Calcific deposits were first seen after six doses of vitamin D, and involved the tubules of the corticomedullary junction. At this time there was marked mitochondrial damage. There was progressive uncoupling of oxidative phosphorylation of the kidney mitochondria, a functional abnormality demonstrable also with magnesium deficiency (Vitale et al., 1957b; Skou, 1962).
13.3. Intensification of Magnesium Deficiency and Renal Damage by Excess Phosphates (Animal)
Diets high in phosphate cause not only bone damage and intensify the cardiovascular lesions of magnesium deficiency but also cause renal damage and calcinosis. Shelling and Asher (1932), who were studying the intensification of vitamin D toxicity by diets high in phosphorus and low in calcium, found that even without any vitamin D supplementation, rats on high P/Ca diets developed hypocalcemia, hyperphosphatemia, and "peppering" of the kidneys with calcium deposits, especially in the corticomedullary zone. When given moderately high vitamin D doses (400 times the antirachitic dose), the rats on high P/Ca intakes developed metastatic calcification and died rapidly, in contrast to the tolerance of much higher doses of vitamin D by rats on a 1:1 P/Ca ratio. Maynard et al.(1958) demonstrated that the severe organ changes of magnesium deficiency reflect imbalance among magnesium, calcium, and phosphorus. The diets that produced the highest blood levels of calcium and phosphorus and the lowest blood levels of magnesium caused the greatest renal damage and calcinosis. Forbes (1963) showed that rats fed diets high in phosphorus but low not only in magnesium but in calcium had the greatest degree of renal calcification, even more than the magnesium-deficient rats fed diets high both in calcium and phosphorus. Dunce et al. (1965) also demonstrated that the renal calcinosis of magnesium-deficient rats was aggravated by increasing the dietary phosphate. They also found that increasing the magnesium intake protected against renal calcification. Spaulding and Walser (1970), concerned about the use of high-dosage phosphate therapy in hypercalcemia, administered amounts of phosphate equivalent to those used clinically to rats with hypercalcemia from hypervitaminosis D. They showed that the phosphate clearly increased calcium deposition in kidneys and heart.
Calves fed a magnesium-deficient diet that was not high in calcium but that was relatively high in phosphorus had renal interstitial fibrosis, with some fibrosis of Bowman's capsule; 7 of the 21 calves had marked tubular necrosis, usually with deposits of calcium (L. A. Moore et al., 1938). Comparable lesions were seen in cows with cardiovascular and articular damage associated with a conditioned magnesium deficiency (Arnold and Fincham, 1950).
The marked susceptibility of a strain of mice with hereditary diabetes to cardiac and renal calcification when fed a diet with a high phosphorus/magnesium ratio (1.2/0.04% of diet), and a phosphorus/calcium ratio of 1, for as little as 10 days (Hamuro et al., 1970) is an intensification and acceleration of the changes that develop later spontaneously in this strain. The degree of calcification was little affected by lowering the calcium intake, but was reduced by increasing the magnesium intake to 0.24% of the diet. It was prevented by increasing the magnesium intake to 0.8% of the diet.
Rats with phosphate-mineralocorticoid-cardiac necrosis also have renal calcinosis, and high dosage magnesium is protective (Selye, 1958a,g).
13.4. Mediation by Secondary Hyperparathyroidism; Protection by Parathyroidectomy
The possible source of the mucoprotein that provides the matrix for calcium deposition in magnesium-deficient animals (Bunce and Bloomer, 1972; Dunce and King, 1976/1980) in bone is suggested by the work of Engel (1952). They showed that administration of parathyroid extract resulted in depolymerization of glycoprotein ground substance of bones and cartilage, and deposition of glycoprotein granules in the renal tubules. Bradford et al. (1962) confirmed that rats given parathyroid extract exhibited deposition of intraluminal glycoprotein material, which preceded calcification. Heaton and Anderson (1965) considered the renal cellular damage and calcification of their magnesium-deficient rats (which were fed a diet containing 590 mg of calcium, 440 mg of phosphorus, and 0.3 mg of magnesium/l00 g to be due to secondary hyperparathyroidism, as a result of the magnesium depletion. Parathyroidectomy prevented the renal calcification caused by magnesium deficiency (Heaton and Anderson, 1965), just as it did that caused by phosphate loading (Clark and Rivera-Cordera, 1972b), which also causes nutritional hyperparathyroidism (Clark and Rivera-Cordera, 1972a; Krook et al., 1975; Review: Clark, 1977). Selye (1958c) showed that PTH, in combination with NaH2PO4 caused intense nephrocalcinosis, as well as cardiovascular and bone damage; magnesium chloride was protective against all three experimental lesions.
13.5. Tissue Magnesium Loss and Damage: Not Parathyroid-Mediated
Exploring the mechanisms by which the phosphate-steroid-cardiorenal damage is experimentally produced, and which was first attributed to mediation by hyperparathyroidism, Lehr (1965b) found that sodium phosphate loading of parathyroidectomized rats caused cardiorenal damage even more rapidly than it did in intact rats. In fact, PTH was protective in this model (Lehr et al., 1967). As his group has demonstrated for the cardiovascular system, the sodium phosphate-loading causes tissue magnesium loss and tissue damage, which precedes the rapid induction of renal calcinosis. These animals succumbed with hypocalcemic tetany, cardiorenal necrosis, and calcinosis. Thus, even though comparable lesions could be produced by calcemic agents such as vitamin D or dihydrotachysterol, in the absence of parathyroid glands, the hypercalcemia was not the cause of the lesions. As is suggested by the magnesium-deficiency studies, soft tissue calcification occurs in damaged recipient sites. Lehr et al. (1966, 1967) concluded that depletion of cellular magnesium, however induced, might be involved in initiation of cellular injury, necrosis of both heart and kidneys following demonstrated sharp drops in magnesium levels in both organs of parathyroidectomized sodium phosphate-loaded rats (Lehr et al., 1966). This pharmacologic model is useful in demonstrating the common denominator in dissimilarly caused cardiorenal damage, cellular magnesium depletion. The nature of the lesions, and the sites at which they occur probably depend upon factors such as concomitant hypercalcemia or hyperphosphatemia, levels of local enzymes or mineralization-inhibitors, and physicochemical factors such as pH and the influence of high concentrations of the minerals involved in precipitation of calcium crystals.
13.6. Phosphatases and Extraskeletal Mineralization
Alkaline and pyrophosphatases have been found, not only in bone, where they function to increase mineralization by breaking down the pyro- and other polyphosphates that inhibit mineralization, but also in normal soft tissues, including the cardiovascular system, liver, brain, and kidneys (Gomori, 1941; Kabat, 1941; Kabat and Furth, 1941; Zetterström, 1951; Kirk, 1959; Kunitz and Robbins, 1966; Romanul and Bannister, 1962) and in urine (Fleisch and Bisaz, 1962b). Avioli et al. (1965) noted that elevated urine pyrophosphate levels characterize rapid bone turn over or breakdown, paralleling hydroxyproline outputs. This compound inhibits crystallization of calcium phosphate as apatites. Thus, an increase in its concentration in urine of patients with osteolysis sheds light on McGeown's (1969) report that the evidence of kidney stones is inversely related to that of osteoporosis.
Low levels of activity of pyro- or alkaline phosphatase should diminish the breakdown of these calcification inhibitors. The inhibition of pyrophosphatase by calcium (Kunitz and Robbins, 1966) might explain the paradoxical finding that there was less calcium deposition in kidneys of magnesium-deficient rats, loaded with phosphate and calcium, than there was in those in which the calcium intake was low (Forbes, 1963). It also helps to understand the protection against renal calcinosis of magnesium-deficient rats by calcium administration (Rayssiguier and Larvor, 1973, 1974); and the observations of Hamuro et al. (1970), who fed a strain of diabetic mice diets with different contents of calcium, phosphorus, and magnesium. The mice low in all these elements had more renal and cardiac calcification than did the magnesium/phosphorus-deficient mice on a normal calcium intake. Whether this reflects calcium inhibition of tissue phosphatase is speculative. In a subsequent study, in which the test mice were fed diets low in magnesium and phosphorus, but adequate in calcium, the plasma alkaline phosphatase levels fell from the high levels seen in control diabetic magnesium-supplemented mice (Hamuro, 1971). At the time the low plasma enzyme levels were obtained, there was renal and cardiac calcinosis, a surprising finding, unless the plasma levels are not indicative of the soft tissue levels. It must be noted that these diabetic mice, which have higher plasma alkaline phosphatase levels on the stock diet than do control nondiabetic mice, spontaneously develop calcinosis, although much more slowly than when they are magnesium depleted.
Manifestly, the degree of stimulation of inhibitors of alkaline or pyrophosphatase levels by high magnesium or calcium levels, respectively, cannot be the entire story. The cellular and membrane damage caused by magnesium depletion allows for an intracellular uptake of excess calcium, with deposition of calcium phosphate (usually amorphous but sometimes crystalline) in the damaged cells. Also, patients with hypercalcemia are prone to metastatic calcification despite pyrophosphatase inhibition by calcium. Formation of calcium pyrophosphate dihydrate crystals, such as have been identified in joints might negate the inhibition by pyrophosphate of calcium salt precipitation, when there is hypercalcemia.
13.7 Magnesium Effect on Precipitation of Calcium Crystals in Urine
There are complex interrelations that determine whether or not urine crystals will form in the renal parenchyma or urine. For example, urine containing pyrophosphate has been shown both to inhibit crystallization of calcium oxalate and hydroxyapatites of calcium phosphate (Fleisch and Bisaz, 1962a; R. G. Russel et al., 1964), and to increase the formation of calcium oxalate (Review: Finlayson, 1974). Mucopolysaccharides have been shown to provide the nidus for calcium precipitation in magnesium deficiency and conditions that enhance osteolysis, and to inhibit aggregation and growth of calcium oxalate crystals (W. G. Robertson et al., 1973). Another inconsistency is the hypercalcemia produced by hypervitaminosis D or hyperparathyroidism, which usually is not associated with urolithiasis. Kushner (1956) noted that both conditions cause increased citrate levels, and concluded that citrate-complexing of urinary calcium functions to prevent urolithiasis. There are many other factors that influence susceptibility to stone formation. Only data directly referable to magnesium are considered, briefly, here.
It has long been known that increased concentration of magnesium in the urine increases the solubility of calcium oxalate (Hammarsten, 1929). Rats fed magnesium-deficient diets, which were rich in oxalates and which produced an alkaline urine, had a high incidence of renal calcification and bladder stones; providing a balanced diet without a high Ca/Mg ratio both prevented stone formation and solubilized some that had been formed (Hammarsten, 1938). Mukai and Howard (1963) showed that addition of magnesium to urine of stone-forming patients blocked the ability of such urine to induce mineralization of collagen in vitro. Administration of about 100 mg of magnesium (as the oxide) three times daily, to 11 patients, with recurrent calcium oxalate crystalluria and stone formation, eliminated the crystal formation, although the oxalate was still being excreted, to a lesser degree. The investigators surmised that the magnesium interfered with formation of the crystals. C. Moore and Bunce (1964) found that administration of 420mg of magnesium oxide daily prevented idiopathic hypercalciuria and stone formation and passage in two subjects within two weeks of starting the treatment. One had formed calcium oxalate stones and one had formed calcium phosphate stones. One had the magnesium therapy discontinued after freedom from calculi for five months, and again began passing stones within two weeks. Prien (1965), on the basis of Gershoff's (1959) work indicating the role of pyridoxine deficiency in oxalate formation, included supplementation with 10 mg of pyridoxine hydrochloride with 4 tablets of magnesium hydroxide (providing about 400 mg Mg/day) in his treatment of calcium oxalate stone-formers. Most of his series of 50 patients showed a marked reduction in formation of new stones. Gershoff and Prien (1967) discussed the mechanisms that might be involved in the increased solubility of the calcium salt excreted, the oxalate of which was only moderately reduced, and the calcium of which was actually increased by 25% in patients treated for a year. Of 36 patients who were observed on treatment for 5 years, 30 had no recurrence or decreased incidence of stone formation. They consider the possibility that increased urinary citrate of magnesium-treated patients (that had been low in the stone formers) might be contributory to the increased solubility of calcium. Melnick et al. (1971/1973; 1971) have reported similarly favorable results among 95 recurrent calcium oxalate stone formers treated with 100 mg of magnesium as the oxide twice daily for two years, and among 47 treated for 4 years. J. Thomas et al. (1978) have demonstrated in vitro and in vivo that magnesium inhibits formation of calcium oxalate crystals. They have obtained the best clinical results with use of magnesium trisilicate, providing 300 mg of magnesium daily. It may be this simple physicochemical effect that is responsible for the difference in incidence of urolithiasis in hard- and soft-water areas (pp. 21-24).
13.8. Clinical Renal Diseases Possibly Related to Magnesium Deficiency
The experimental evidence that magnesium deficiency during pregnancy produces greater fetal than maternal magnesium deficiency raises the possibility that renal tubular abnormalities, such as are produced in weanling magnesium-deficient animals, might occur in utero. No studies of the renal structure of fetuses of experimental magnesium-deficient mothers have been found, and thus this possibility remains speculative. Microscopic examination of kidneys of stillborn babies of mothers subject to magnesium deficiency should provide valuable data.
After birth, there are several conditions that lead to magnesium deficiency, both in the neonatal period and later in infancy. Infants who do not survive neonatal asphyxia or sodium bicarbonate or sodium lactate treatment of their neonatal or postoperative acidosis, both anoxia and acidosis causing egress of magnesium from the cells, should have their renal parenchyma carefully studied, especially for evidence of tubular cellular and subcellular damage. The kidneys of erythroblastotic infants failing to survive exchange transfusion with citrated blood (which chelates magnesium) should be similarly examined. Definitive data can be obtained from experimental models of these perinatal abnormalities, which can provide electron microscopic evidence of very early renal changes, as well as light microscopic evidence of sequellae. Such studies should include evaluation, not only of kidneys, but of cardiovascular tissues (especially intramural coronary arteries, myocardium, and endocardium) and bone.
Because neonatal hypocalcemia is usually noted first, and usually aggressively treated with calcemic agents to control the neuromuscular irritability and convulsions, what might be an underlying magnesium deficiency is usually detected only by the time magnesium depletion has developed to the point of severe hypomagnesemia. Familial magnesium malabsorption might be a contributory factor in infants and children with the most severe manifestations. Neonatal magnesium deficiency and hypoparathyroidism secondary to gestational magnesium deficiency in some instances, and to high phosphate + vitamin D intakes in others, is likely to contribute to less severe but possibly damaging tissue magnesium loss. The renal damage of such treatment might lead to long-term intensification of magnesium deficiency by causing damage to the portion of the renal tubules where active magnesium reabsorption occurs.
13.8.1. Renal Tubular Defects in Magnesium Reabsorption
As noted earlier, most active renal tubular reabsorption of magnesium occurs in the ascending limb of the loop of Henle (ALLH), which provides insight into clinical magnesium wastage, most early experimental magnesium deficient renal damage occurring in the tubular cells of the corticomedullary area, with microliths of the loop of Henle, convoluted and distal tubules, and with damage to ALLH. That these experimental findings are relevant to the clinical situation is suggested by fragmentary findings.
Whether magnesium deficiency contributes to clinical aminoaciduria, as it does the experimental model should be investigated. Its occurrence in renal tubular acidosis, vitamin-D-resistant rickets, and hyperreactivity to vitamin D, in all of which conditions magnesium deficiency might play a role, is suggestive.
13.8.1.1. Contributions to Clinical Renal Magnesium Wastage by Calcemic Factors and Phosphate Therapy
Calcium deposits in the lumens of the proximal renal tubules and of the ALLH have been described in an infant whose symptomatic hypocalcemia had been unsuccessfully treated with calcium infusions and high-dosage vitamin D, before severe magnesium deficiency was detected in a specimen taken the last day of life (Vainsel et al., 1970). Another infant, whose persistent infantile hypocalcemia had been treated by intensive calcium therapy and then, when complicated by intractable diarrhea, with addition of high-dosage vitamin D (10,000 IU/day) was then found to have hypomagnesemia and tubular acidosis. At autopsy there were calcium deposits in the distal tubules and collecting ducts. No parathyroid tissue was found at autopsy (Taitz et al., 1966). Infants with hypervitaminosis D and infantile hypercalcemia, which seems to be caused by hyperreactivity to vitamin D (Review: Seelig, 1969b) have also been found to have intraluminal calcium deposits predominantly in the outer half of the renal medulla (Dawson et al., 1954; Rhaney and Mitchell, 1956). Nephrocalcinosis infantum, that Lightwood (1935) first associated with renal tubular acidosis, hypophosphatasia, hyperoxalemia, and sarcoidosis (which is associated with hyperreactivity to vitamin D) is also associated with renal tubular lesions and calcium deposits involving Henle's loop and tubules immediately proximal and distal to it (J. A. James, 1956; Kushner, 1956; Shanks and MacDonald, 1959; T. Ferris et al., 1961; Paunier et al., 1968a). Hyperreactivity to vitamin D, of infants who were receiving excessively fortified milk and infant foods was implicated in Great Britain in renal tubular acidosis (Fig. 13-2)(Lightwood and Butler, 1963) and in nephrocalcinosis (Shanks and MacDonald, 1959). Development of such abnormalities in children who were being given high-dosage vitamin D for vitamin D-refractory rickets (with or without aminoaciduria) or for idiopathic hypoparathyroidism and hypocalcemia (T. Ferris et al., 1961; Paunier et al., 1968a; Moncrieff and Chance, 1969) suggests that magnesium deficiency might have played a contributory role, first to the vitamin-D-refractory rickets or hypocalcemia and then to the increased susceptibility to nephrotoxicity of vitamin D. It seems plausible that the presenting tetany and convulsions of the babies with hypocalcemia, renal tubular acidosis, and nephrocalcinosis (J. James, 1956; Ferris et al., 1961) might have been contributed to by magnesium deficiency. Familial renal tubular wasting of magnesium has been reported in siblings with renal tubular acidosis, renal calcinosis, and hypocalcemia that was resistant to very high doses of vitamin D (Michelis et al., 1972). The older child (a 10-year-old girl) had active rickets; the bone age of the younger brother (6 years old) was three standard deviations below normal. Neither child's hypomagnesemia (1.1, 1.2 mEq/liter) responded to high- dosage oral magnesium supplementation, a failure found due to renal wastage rather than malabsorption of magnesium. It is conceivable that the initiating abnormality in these children might have been hyperreactivity to vitamin D, which might have led to hypercalcemia and ALLH damage, that was responsible for their persistent renal magnesium wastage. It is provocative, however, that both children had been born prematurely. The older child was jaundiced at birth and developed a convulsive disorder at the age of 6. The younger child underwent surgery at 6 weeks of age. The premature births and complicated neonatal courses might have contributed to early magnesium deficiency that might have contributed to hyperreactivity to (toxic effects of) vitamin D. The older child had had polyuria from the age of two; the younger child from the age of 1.
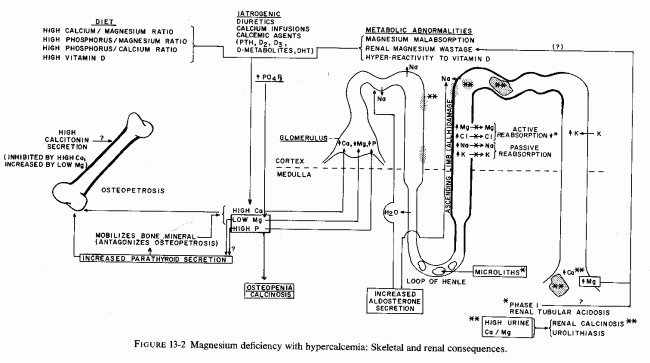{kind=link}
Another child with vitamin-D-resistant rickets was found to have hypomagnesemia (0.7 mEq/liter) and two times the normal renal clearance of magnesium when he was ten years old, following two years of very high (15 mg/day) vitamin D dosage (Sann et al., 1975). He had manifestations similar to those seen in infantile hypercalcemia during the first year of life (anorexia and insomnia), and he exhibited poor growth. He had clear evidence of kidney disease from two years of age, at which time he developed proteinuria; by six he had polyuria and polydipsia and marginally high serum calcium. By eight he had skeletal demineralization, for which the high-dosage vitamin D was prescribed. Renal biopsy showed juxta-glomerular hyperplasia, and he had hyperrenism and normotensive aldosteronism. At this time, sodium restriction caused hyponatremia. Angiotensin-infusion did not raise his blood pressure, and he was judged to have a form of Bartter's syndrome (Bartter, 1962). A second renal biopsy confirmed the juxta-glomerular hyperplasia and revealed proliferative endarteritis of the efferent arterioles. Two years later, the osteoporosis had progressed, and his phosphaturia, magnesiuria, and hypophosphatemia and hypomagnesemia were identified and found to be refractory to the potassium-sparing diuretic (triamterene) that was given in the hope that it might spare magnesium. It is provocative that hypomagnesemia has also been identified in Bartter's syndrome (Brackett et al., 1968; Sutherland et al., 1970; Mace et al., 1973), a condition that Kurtzman and Gutierrez (1975) suggest may be a syndrome caused by ALLH dysfunction.
Reviews of the literature on renal damage caused by hypercalcemia-inducing agents such as vitamin D excess (Epstein, 1960) and hyperparathyroidism (Pyrah et al., 1966) show that the earliest lesions are in the loop of Henle, the ALLH, and the distal convoluted tubules and collecting tubules and that the duration of the hypercalcemia can be as short as one to three days to produce significant damage. The damage becomes irreversible with sustained hypercalcemia. That such lesions can cause renal magnesium loss is suggested by the work of Massry et al. (1967), who reported an acquired defect in renal tubular reabsorption of magnesium in a 44-year- old woman with surgical hypoparathyroidism. They considered the renal defect as one likely to have been caused by hypercalcemia, subsequent to long-term treatment with vitamin D and thiazide diuretics (each of which favors retention of calcium over magnesium). Hypomagnesemia was detected (0.9-1.2 mEq/liter), which was not corrected by withdrawal of the diuretic and the vitamin D. Her high renal clearance of magnesium persisted, and did not change on administration of PTH. Hypercalcemia of immobilization of an adolescent boy, who had suffered multiple fractures, also resulted in sustained increased renal clearance of magnesium (Hyman et al., 1972).
It is possible that early renal tubular damage, caused by calcemic agents (supra vide) or by phosphate therapy that causes soft tissue calcinosis, including renal tubular damage (Bulger et al., 1930; Albright et al., 1932; Carey et al., 1968; Marti and Cox, 1970; Dudley and Blackburn, 1970) can be reversed with adequate magnesium repletion, once the hypercalcemia has been controlled. A clue that this may be so derives from the observations of Dooling and Stern (1967), who studied the magnesium status of a six-day-old infant whose hypocalcemic convulsions had been intensively treated with oral and parenteral calcium. (It should be kept in mind that such infants are also generally hyperphosphatemic. The baby excreted large amounts of magnesium (16.3 mg/kg/24 hr) while he was hypomagnesemic (0.6-0.7 mEq/liter) and being treated by intramuscular administration of 0.25 ml of 50% magnesium sulfate every 6 hours (25 mEq M2+/24 hr). His urinary magnesium output did not fall until the magnesium dosage was doubled. By the fourth day of high- dosage magnesium, with resultant stable elevation of his serum magnesium level at 1.6 mEq/liter, his urinary magnesium output had fallen to 3.06 mg/kg/24 hr, a fivefold drop in daily urinary magnesium output. It is tempting to speculate that microliths might have formed in the loops of Henle, with resultant early damage to the broad ascending limb when he was being loaded with calcium and was magnesium depleted. Early during the magnesium supplementation, there might have been impaired tubular reabsorption of magnesium. When the amount of magnesium given was increased, solubilization of presumed calcium microliths and tubular cells might have taken place, with resultant increased active tubular reabsorption of magnesium.
13.8.1.2. Contribution to Clinical Renal Magnesium Wastage by Malabsorption
Specific genetic magnesium malabsorption or general intestinal malabsorption might cause severe enough magnesium depletion to cause renal tubular damage directly, or as a consequence of calcemic therapy of secondary hypocalcemia. Direct evidence that this might be so has recently been provided by Rapado and his colleagues (Rapado et al., 1975; Rapado and Castrillo, 1976/1980a). They reported a 12-year-old child with a diagnosis of nephrocalcinosis from early life, who developed overt rickets when her hypercalciuria was treated, first with sodium cellulose phosphate and then with hydrochlorthiazide. She was then found to have hypomagnesemia (0.5 mEq/liter) and required parenteral magnesium therapy because she was both a magnesium malabsorber and renal waster. Of two additional patients with nephrocalcinosis, one young man with latent tetany of hypomagnesemia (0.75 mEq/liter) and hypocalciuria (6.8 mg/100 ml), reported by this group, was a renal magnesium waster. Their third patient with hypomagnesemic (0.65 mEq/liter) nephrocalcinosis was a young man whose urinary magnesium levels did not decrease on a low-magnesium intake, and whose fecal excretion of magnesium was higher than his intake, suggesting magnesium malabsorption. This patient's serum calcium level was only marginally low (8.3 mg/100 ml), and he had tachycardia and hypertension. All three patients had hypercalciuria, and their urinary calcium output increased with magnesium therapy.
The patient reported by Freeman and Pearson (1966) as a renal tubular waster of magnesium, had had a history suggestive of steatorrhea and growth failure during her first year of life, which suggested early magnesium depletion. She came from a family with a high incidence of hypomagnesemia: One of her sons had serum magnesium levels of 1.12, 1.20 mEq/liter on two occasions; another son, a sister, a maternal cousin, a daughter, and the patient's mother had marginally low serum Mg levels (1.50, 1.47, 1.68, 1.51, and 1.70 mEq/liter, respectively, strongly suggestive of a genetic trait. Whether the inherited trait was primary renal dysfunction or magnesium malabsorption is not clear. Another patient, one who had had gastrointestinal disease (ascariasis, followed by gastroenteritis) before she was two years old, exhibited subsequent growth failure, intermittent glycosuria and aminoaciduria, and X-rays suggestive of osteoporosis (B. Booth and Johanson, 1974). The boy, whose hypomagnesemia was not detected until he was six years of age (Miller, 1944), might have had infantile hypomagnesemia, as suggested by his long history of neuromuscular irritability. His osteochondritis of three years duration, resembling that of a five-year-old boy with renal magnesium wasting (Klingberg, 1970), suggested to Booth and Johanson (1974) that the child reported by J. F. Miller (1944) might also have had a renal tubular defect. It should be noted, however, that the defect might as readily have been in intestinal absorption of magnesium
Perhaps the first recorded instances of excessive urinary output of magnesium despite marked hypomagnesemia were two patients with peptic ulcers, one with prolonged nasogastric suction (who undoubtedly was not absorbing normal amounts of magnesium) and another who had had several complicated surgical procedures (Martin et al., 1952). That surgical patients do not conserve magnesium efficiently during the early postoperative days has been shown repeatedly. Data are generally not available on the nature of the antacids taken, prior to the suction or surgery, but if they were calcium rather than magnesium preparations, the patients might have had a relatively high Ca/Mg dietary ratio prior to the acute situation that intensified their magnesium loss
Note should be taken of the incomplete distal renal tubular acidosis seen in two women who developed hypomagnesemic hypocalcemia as a result of intestinal malabsorption: one following intestinal resection for regional enteritis, the other with nontropical sprue (Passer, 1976). Since these patients had hyperparathyroidism (by immunoassay) and treatment with magnesium alone corrected the abnormal renal function, the author speculated that the resistance to the calcemic effects of the endogenous PTH might be related to abnormalities in vitamin D metabolism, which was corrected by the magnesium.
13.8.1.3. Miscellaneous Factors in Renal Magnesium Wastage
Randall et al. (1959) was the first to propose a functional renal tubular defect as an explanation for the failure of a 38-year-old man with mild diabetes mellitus, pyelonephritis, focal seizures, and electrocardiographic abnormalities to conserve magnesium. Before dying with extensive arteriosclerosis and myocardial infarct, he had exhibited hypokalemic alkalosis, hypocalcemia, and hypophosphatemia. They noted that additional patients with renal tubular disease wasted magnesium. Two adult sisters and an unrelated 22-year-old woman were first identified as having impaired renal conservation of magnesium and potassium, in association with metabolic alkalosis; one also had hypochloremia (Gitelman et al., 1966a). The sisters had dermatologic manifestations resembling those seen in magnesium-deficient animals; the third patient had recurrent carpopedal spasm. All exhibited slightly elevated aldosterone secretion without hypertension and had minor ECG abnormalities; two had muscle weakness.
Chronic hypomagnesemia and recurrent episodes of neuromuscular irritability and severe abdominal pain have recently been attributed to a renal tubular defect in magnesium reabsorption in a boy, whose mother also has had subnormal serum Mg levels (Paunier and Sizonenko, 1976/1980). As in the previously reported patients with this renal defect, even supplementation with large doses of magnesium failed to normalize the serum magnesium levels.
The oldest patient, at the time of first detection of renal wastage of magnesium, is a postmenopausal woman who developed normocalcemic latent tetany of marginal deficiency several years after total hysterectomy (Seelig et al., 1975). She has a less severe form of renal Mg wasting and less marked hypomagnesemia than the other cited patients. Her condition is associated with normotensive, intermittent aldosteronism and increased plasma renin activity (PRA), only manifest in response to dietary Mg restriction or to hormonal challenge (i.e., deoxycorticosterone acetate) that increased her magnesium deficit (Seelig et al., 1976/80). Since she has also had hypochloremia, it has been proposed that she has malfunction of the ALLH, where not only magnesium but chloride is actively reabsorbed (Rocha and Kokko, 1973; Burg and Green, 1973; Kurtzman and Gutierrez, 1975). It seems plausible that her renal dysfunction and concomitant abnormalities-sodium retention, peripheral edema, hypokalemia responsive to magnesium, hypercapneic alkalosis, and hormonal aberrations-may be the result of magnesium insufficiency, since all of these findings have been reported in experimental magnesium deficiency (Review: Seelig et al., 1976/1980; Whang and Welt, 1963; Ginnet al., 1967; Cantin, 1970; Elin et al., l97la; El Shahawy, 1971; Cantin and Huet, 1973).
13.8.2. Renal Damage during Pregnancy: Related to Magnesium Deficiency?
Magnesium deficiency has been implicated in preeclampsia and eclampsia. Possibly it contributes to the renal damage of eclampsia. The involvement of the small (coronary) arteries in magnesium deficiency makes one suspicious that the renal arteriolar disease of young toxemic primiparas (Smythe et al., 1964), who are particularly prone to magnesium deficiency of pregnancy, might also be a consequence of magnesium inadequacy. DeAlvarez and Gabrio (1953) implicated arteriolar spasm in the decreased glomerular filtration rate of patients with toxemias of pregnancy.
The attempts to counter the leg cramps of pregnancy (which might be contributed to by magnesium deficiency) by calcemic therapy, might intensify the magnesium deficiency directly and as a result of damage to renal tubular cells (supra vide). The resultant high calcium/magnesium ratio particularly in arterial tissue [such as has been implicated in increased arterial tension (Review: Haddy and Seelig, 1976/ 1980)] might similarly be a factor in the hypertension of abnormal pregnancy. Calcemic supplements to magnesium-deficient pregnant women might contribute to urinary calculi of pregnancy, which has reported in 0.05-0.35% of pregnancies. (McVann, 1964; R. Harris and Dunnihoo, 1967). Since estrogen lowers the urinary content of calcium and raises its citrate level (Shorr, 1945), both effects that militate against calcium stone formation, the degree of magnesium deficiency might well be fairly profound for calcareous stones to form during pregnancy. On the other hand, the resultant hyperparathyroidism of pregnancy might directly increase the propensity toward renal calcinosis formation, as well as hypertension, both being consequences of hyperparathyroidism (Review: Pyrah et al., 1966).
Only brief reference will be made here to the speculation that magnesium deficiency might be contributory to proliferative arteriolar sclerosis, found in the kidneys as well as in other tissues, including the myocardium. As in experimental magnesium deficiency, in which there is arteriolar disease with subendothelial, muscle wall, and endothelial proliferation, with increased wall thickness/lumen ratio, renal (and other) arterioles have subintimal and medial abnormalities with encroachment on the lumen (Review: Ditzel, 1954). The lesions are not identical pathologically and the glomerular capillary changes of the Kimmelstiel-Wilson lesion have not been described in magnesium deficiency, but loss of magnesium by diabetic patients raises the possibility that the arterial changes might have a component contributed to by magnesium deficiency.
Part III: Chapter 14
SKELETAL AND RENAL EFFECTS OF MAGNESIUM DEFICIENCY
The possibility that magnesium deficiency might be contributory to, or might accompany, the abnormalities that cause osteopenia, hypocalcemia, hypercalcemia, and renal and cardiovascular disease is rarely considered in initiating therapy. Refractoriness to direct attempts to correct hypocalcemia and hypokalemia are now increasingly leading to investigation of serum magnesium levels, and less frequently to other (better) means of ascertaining the body's magnesium status. Emphasis is placed, in this chapter, on the problems that can result from treatment of either hypo- or hypercalcemia by agents that cause magnesium loss, when the primary disorder is one resulting in magnesium deficiency. Accepting the difficulties in evaluating the magnesium status, it is proposed that serum magnesium levels and 24-hour urinary magnesium outputs be made part of the routine initial diagnostic program. Since higher serum levels of magnesium are tolerable without serious hazard, except perhaps when there is hypercalcemia, it is suggested that magnesium therapy be tried before calcium loading of patients who have disorders that might make them susceptible to magnesium deficiency.
It is usually recommended that pregnant women drink ample amounts of milk (which in most industrialized countries is "fortified" with antirachitic amounts of vitamin D) and take vitamin supplements that also provide an antirachitic dose of vitamin D. As pointed out earlier, the magnesium intake is likely to be meager. Then, when leg cramps of pregnancy develop (which can be caused by magnesium deficiency and hypomagnesemic hypocalcemia), the usual therapeutic approach is generally administration of calcium. Rarely is the magnesium status investigated, and magnesium treatment tried. There have been publications, however, that have shown that, both in normal and abnormal pregnancy, serum levels of magnesium tend to be low, even when corrected for hemodilution. Metabolic balance studies have shown that normal pregnant women should ingest sufficient magnesium to maintain a strongly positive balance, to meet both her needs and those of the fetus. It has been proposed that some of the abnormalities of pregnancy might be a consequence of magnesium deficiency. The fetus might be at even greater risk, experimental gestational magnesium deprivation causing greater fetal than maternal magnesium deficiency. Factors that increase magnesium requirements, such as gestational hypervitaminosis D, have caused congenital cardiovascular, renal, and skeletal defects. Suggestive evidence has been presented, and a theory promulgated that magnesium deficiency during pregnancy might be contributory to several "congenital" abnormalities of the heart, arteries, kidneys, and bones. Cardiac outflow abnormalities (such as can be produced by experimental hypervitaminosis D) have been found in conjunction with endocardial fibroelastosis. Infantile coronary or generalized arteriosclerosis, cardiomyopathy, and dysrhythmias, sometimes leading to sudden death, are seen alone or in combination with gross cardiac abnormalities. Such infants often have renal calcinosis, and if they survive the early months often have growth and mental retardation. Osteogenesis imperfecta, which resembles lesions that have been produced in pups of vitamin-D-poisoned rats, also resembles lesions of severe congenital hypophosphatasia, and is sometimes accompanied by cardiac and renal abnormalities, such as are seen in hypervitaminosis D. Neonatal hypoparathyroidism is common, and must be attributable to influences in utero, speculated to be gestational magnesium deficiency. One may wonder whether more intense magnesium depletion might so suppress the parathyroids in utero as to be responsible for congenitally deficient parathyroid tissue: "idiopathic" primary hypoparathyroidism. When the diseases are severe or familial, the patient's and the mother's intestinal absorption and renal tubular reabsorption of magnesium should be explored, as should that of other close relatives; since familial defects of magnesium metabolism have been recognized, it is conceivable that this might be a flaw that intensifies lesions caused by other heritable disorders, or even underlies some of them.
Them is a wide spread of vitamin D requirements and susceptibility to its toxicity. Thus, the practice of routinely providing much more than prophylactic amounts of vitamin D during pregnancy should be reevaluated, taking into account the usually high dietary intakes of phosphate, which, like low-magnesium intakes, intensifies vitamin D toxicity. Requiring investigation is the influence of such imbalances on the maternal organism and the placenta, and systematic investigation of the fetal organs and bones should be undertaken of stillborn infants, and of experimental models. Until definitive experimental data are available, we should keep in mind that magnesium has protected against experimental vitamin D and phosphate toxicity, and that magnesium deficiency (as is likely during pregnancy, especially in immature mothers and in women who have had frequent pregnancies, but also in less stressed mothers) has intensified the lesions of hypervitaminosis D and phosphate loads. Thus, magnesium supplementation (to a total of at least 7-10 mg/kg/ day) is suggested as a minimum for those without magnesium malabsorption or renal wastage. If either of those abnormalities of magnesium metabolism is detected 'and it should be sought if there is a familial history of suspect abnormalities), the magnesium supplementation should be correspondingly higher, and might have to be parenterally given.
Considered in detail are the risks of treating neonatal hypocalcemia, which might well be a consequence of magnesium depletion, with calcemic agents. Failure of response of neuromuscular irritability is often the first clue to the necessity of evaluating the magnesium status, and favorable response to magnesium therapy the proof. However, during the time that a magnesium-deficient infant is being loaded with calcium, or given agents that cause bone resorption, damage can be inflicted on the heart, arteries, kidneys, and bones, and (especially in babies with genetic susceptibility to abnormalities of these tissues) permanent lesions might result. For example, magnesium deficiency and vitamin D excess each causes lipid abnormalities and damage to small and large arteries, respectively. In addition, the early renal lesions of magnesium deficiency (in the face of calcemic factors) are in the tubules, in the area of active magnesium reabsorption. Thus, such therapy might be contributory to establishment of transitory or permanent renal magnesium wasting. With continued calcemic treatment (or dietary custom that provides only moderate excess of vitamin D to infants who are hyperreactive to vitamin D, are magnesium deficient, or both), cardiac outflow abnormalities, endocardial fibroelastosis, premature atherosclerosis, renal calcinosis, and osteosclerosis as well as mental retardation, might result.
The use of high-dosage vitamin D or its derivatives in the treatment of refractory osteopenias might similarly result in cardiovascular and renal damage, other soft tissue calcinosis, and osteosclerosis, rather than normal bone, which requires optimal magnesium for normal osteocyte activity and matrix formation. Little has yet been done to correlate the osteopenia or brittle chalky bones produced by either experimental magnesium deficiency or by vitamin D excess, the degree depending on the amount of calcium and phosphate in the diet. As regards the use of high-dosage calcemic agents for postmenopausal osteoporosis, reference should be made to the estrogen/parathyroid/magnesium interrelationships that suggest that magnesium's effect on osteocytes and matrix formation might find applicability in preventing further loss, if not serving to increase formation of organic matrix.
Inadvertent proof was provided that hypervitaminosis D produces metastatic calcification when very high doses of vitamin D were used to treat arthritis, even when the intake of calcium was not high (Danowski et al., 1945; Mulligan, 1947; Frost et al., 1947; Howard and Meyer, 1948; Reed, 1950; Christensen et al., 1951; Verner et al., 1958). In such instances, the calcium, phosphate, and matrix were drawn from the skeleton and deposited in soft tissues. In one of the studies (Frost et al., 1947) magnesium was studied and found to be low during the vitamin-D-toxic period and to rise when the overdosage was stopped. The evidence that some arthritic processes might be consequences of magnesium depletion suggests that seeking and correcting magnesium deficiency might be useful.
It is advisable to explore the magnesium status of patients with osteopenias before loading them with calcemic agents, which might prove useless in some or unduly toxic in others if magnesium deficiency is present. If hypercalcemia has already been induced by high doses of such agents as vitamin D or its congeners or metabolites, or by parenteral loads of calcium, the magnesium serum level and 24- hour urinary output should be determined. A parenteral magnesium load may be inadvisable until the hypercalcemia is corrected, and not by phosphate loading.
Because hypercalcemic crises are life-threatening, emergency treatment is directed toward lowering the circulating calcium levels quickly, by hydration with saline or dextrose in water, and increasing its urinary excretion with a potent diuretic such as furosemide, by administration of phosphate to increase its precipitation, hopefully in the bones, and by agents such as calcitonin to shift the calcium to bone, or mithramycin to antagonize bone resorption (Newmark and Himathongkam, 1974). Corticosteroids, which act more slowly, are recommended in long-term control of chronic hypercalcemia. Unfortunately, saline and furosemide diuresis, phosphate loads, and corticosteroids all increase magnesium loss, which is also caused by the hypercalcemia as well as frequently by the diseases that caused the hypercalcemia in the first place. Furthermore, inorganic phosphates have resulted in ectopic, sometimes fatal calcification (infra vide).
Hydration and furosemide diuresis are acceptable, until calcitonin can be obtained. Calcitonin is a preferable agent because it increases deposition of calcium in bone, stimulating bone alkaline pyrophosphatase (Orimo et al., 1970), without transferring calcium to soft tissue sites (Chausmer et al., 1965). In fact, there have even been reports that calcitonin protects against soft tissue calcification (Gudmundsson et al., 1966; Kenny and Heiskell, 1965; Gabbiani et al., 1968; Rasmussen and Tenenhouse, 1967; Rayssiguier and Larvor, 1974a). Once the plasma calcium levels are lowered, magnesium therapy can be substituted for the calcitonin, evidence having been obtained that calcitonin secretion is stimulated by increased magnesium (Radde et al., 1970; Bell and Kimble, 1970; Care et al., 1971; Littledike, 1970; Littledike and Arnaud, 1971; S. P. Nielsen, 1974). Additionally, moderately increased magnesium levels suppress parathyroid secretion (Care et al., 1966; Buckle et al., 1968; Gitelman et al., 1968a; Massry et al., 1970b; Sherwood, 1970; Sherwoodet al., 1970; Altenahr and Leonhardt, 1972). Competition between calcium and magnesium for a common renal tubular reabsorptive pathway (Samiy et al., 1960a,b; Charbon and Hoekstra, 1962; Ardill et al., 1962; Heaton et al., 1964; Massry and Coburn, 1973) has also been credited for the increased urinary excretion of calcium and drops in serum calcium that accompany magnesium loads (Womersley, 1956; Chesley and Tepper, 1958; Kelly et al., 1960; Kemeny et al., 1961: S. P. Nielsen, 1970; Nielsen and Jorgensen, 1972).
It is recommended that magnesium not be given until the acute hypercalcemia been lowered, intensification of soft-tissue calcinosis having been produced by magnesium given to rats with experimental hypercalcemia caused by hypervitaminosis D (Whittier and Freeman, 1971).
14.4.1 Risks of Phosphate Therapy
Inorganic phosphate therapy has been utilized and warned against for many years in the treatment of hypercalcemia and of skeletorenal disorders. Oral administration of inorganic phosphates was found, almost 50 years ago, to reduce the acute hypercalcemia of patients with hyperparathyroidism (Bulgeret al., 1930; Albright et al., 1932). However, both groups of investigators expressed concern about the risk of promoting nephrolithiasis or other extraskeletal calcification. Bulger et al. (1930), for example, found extensive calcification of lungs, gastric mucosa, and kidneys when a patient died of bronchopneumonia a few days after the infusion. Shortly thereafter, Bulger and Gausman (1933) demonstrated that hyperparathyroidism causes negative magnesium balance. In 1962, Dent reintroduced phosphate therapy for hypercalcemia. One of his patients responded well; the other developed extensive painful ectopic calcification. Four years later, R. S. Goldsmith and Ingbar (1966) again described the usefulness of phosphate loads for treatment of life-threatening hypercalcemia, applying it also to patients with neoplasms. They obtained rapid and dramatic decreases of serum calcium levels and improvement of symptoms in 16 of their 20 patients. Ten died, of whom 7 had autopsies. Five had extraskeletal calcification. One, who had not been examined postmortem, had died of a massive infarction the day after the phosphate infusion. Because of uncertainty that these six instances were related to the treatment, and of the rapidity with which the phosphate lowered the plasma calcium level, R. S. Goldsmith (1970) reiterated his recommendation that this approach was most va1uable for hypercalcemic crisis in his critical review of Eisenberg's (1970) caution as to the risk of producing metastatic calcification. Eisenberg (1970) noted the instances in which such calcification had been reported after either intravenous or oral administration of large doses of phosphates, and warned of the likelihood that the calcium would precipitate out in soft tissues. For example, Schackney and Hasson (1967) reported hypotension and acute renal failure in two patients whose hypercalcemia had been treated by phosphate infusions. One had extensive metastatic calcification in the heart, lungs, kidneys, and pancreas; the other exhibited no metastatic calcification on autopsy. Breuer and LeBauer (1967) reported a patient with multiple myeloma and hypercalcemia, who had a good temporary clinical and chemical response to intravenous and oral phosphate treatment, but who suddenly died with renal insufficiency and pneumonia and was found to have extensive pulmonary and renal calcification. Carey et al. (1968) reported metastatic calcification involving the endocardium, coronary arteries, and kidneys (glomerular, intraluminal, and interstitial) in a patient whose hypercalcemia of neoplastic origin had been treated with inorganic phosphate infusions. Marti and Cox (1970) reported additional patients who developed irreversible calcinosis, particularly of renal tubules and lungs, following phosphate infusions for hypercalcemia resulting from bone metastases. Dudley and Blackburn (1970) recommended slit lamp conjunctival examination to detect early extraskeletal calcification, such as they found in seven of nine patients who had been treated with high-dosage oral phosphates. Five had been treated for hypercalcemia; two with hyperparathyroidism developed impaired renal function during therapy. Of four normocalcemic patients, who were being given phosphate therapy for renal calculi, three developed conjunctival calcification, one developed radiologic evidence of renal and one of arterial calcification.
Thus, although inorganic phosphate has been effective in reducing hypercalcemia and the incidence of calcific urinary stones, it carries the risk of soft-tissue calcinosis, such as is seen with magnesium deficiency, and is intensified by phosphate loading. Monsaingeon et al. (1971/1973) found that oral inorganic phosphate loads (2.25 g/day) decreased the urinary magnesium concentration more than it did that of calcium in 70% of 29 patients with urinary calculi. They cautioned that it is necessary to monitor magnesium levels in patients treated with phosphates.
Short-term administration of cellulose phosphate to normal subjects has reduced the intestinal absorption and urinary output of both calcium and magnesium (Dent et al., 1964), and caused a gradual decline in serum magnesium (but not calcium) levels (Parfitt, 1975). That long-term administration of cellulose phosphate, given to reduce the urinary calcium in stone formers, can cause magnesium depletion is indicated by Sutton's (1968) study. He reported hypomagnesiuria and hypomagnesemia in a recurrent stone former, who had been treated with a low calcium diet and oral cellulose phosphate for 6 years. His plasma magnesium remained between 0.65 and 1.25 mEq/liter and his 24-hour urinary magnesium between 2 and 15 mg during a year of observation, while he was on that regimen.
Other phosphates have induced fewer problems, possibly in part because of lesser depletion of magnesium, and in part because of their increase in urinary output of inhibitors of calcium crystallization in urine. For example, orthophosphate (disodium hydrogen phosphate dihydrate) administration (12 g/day = 1.98 g P/day) to patients with recurrent renal calculi, to reduce the intestinal absorption of calcium and thereby to reduce urinary calcium excretion, was found also to increase the urine citrate and pyrophosphate levels, but to influence the magnesium levels and balance only slightly. It caused a more profoundly negative calcium balance, decreased urinary calcium output, but caused a net increase in the Ca/Mg urinary ratio. The crystallizing propensity was reduced, probably largely because of the orthophosphate-induced pyrophosphate and citrate levels. The investigators who showed that inorganic pyrophosphate inhibits the precipitation of hydroxyapatite crystals in vitro (Fleisch and Neuman, 1961) developed condensed phosphates (diphosphonates) that are less readily hydrolyzed and that are effective in preventing ectopic calcification in vivo (Irving et al., 1966; Francis et al., 1969). A diphosphonate, which is under investigation for its inhibition of ectopic calcification and of bone resorption (Francis et al., 1969; Fleisch et al., 1969; Russell et al., 1971; Michael et al., 1971; Saville and Heaney, 1972), has been shown to cause negative magnesium balance on long-term in children with ectopic calcification (Uttley et al., 1975).
With so much evidence that magnesium deficiency accompanies hypercalcemia and its treatment, it is tempting to recommend prompt magnesium repletion. However, one must keep in mind the magnesium dependence of phosphatases that destroy the polyphosphates (including the pyrophosphates) that inhibit precipitation of calcium salts in the soft tissues. In fact, several of the British investigators, who reported the severe form of infantile hypercalcemia, suspected that use of magnesium laxatives might have intensified the syndrome that is characterized by renal, cardiovascular, and brain damage and calcinosis. Thus, as indicated earlier, serum calcium levels should be lowered first, by the least dangerous means, before instituting magnesium therapy. Without hypercalcemia or hyperphosphatemia, magnesium activation of soft-tissue alkaline or pyrophosphatase should not present a danger of precipitation of calcium phosphate salts. Magnesium stimulation of bone alkaline or pyrophosphatase should function to take up calcium and phosphate, particularly if the magnesium suppresses parathyroid and increases calcitonin secretion.
Among the tissues damaged by magnesium deficiency, those of the cardiovascular and skeletal and the urinary tract are listed on Table 14-1. Some of the abnormalities cited in several of the diseases to which there is reason to believe magnesium deficiency is contributory are disorders that are comparable to those seen in experimental magnesium deficiency. Unfortunately, some of the early findings (such as hypocalcemia, neuromuscular irritability, and osteopenias) suggest direct treatment with the obviously deficient substance, calcium, or by agents that normally function to increase calcium absorption and its blood levels. When the hypocalcemia or osteopenia is secondary to magnesium depletion, such treatment can intensify the magnesium loss, increase the cellular damage (caused by magnesium deficiency), and lead to metastatic calcinosis (Figure 14-1).
{kind=link}
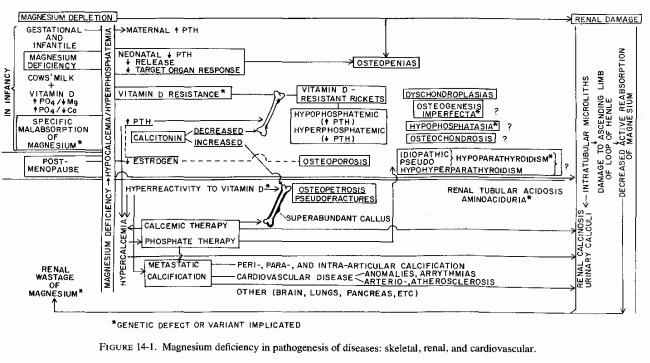{kind=link}
Thus, the underlying abnormality (metabolic or dietary or both) that prevents normal magnesium utilization can lead to abnormal function or response to parathyroid hormone, vitamin D, or calcitonin, with possible production of a variety of osteopenias. Most of the diseases have their roots during gestation or early infancy. One, postmenopausal osteoporosis, is entered because of the possibility that the drop in estrogen secretion might contribute to relative hyperparathyroidism. High dietary phosphate intakes, which can contribute to major disorders of infancy, might also play a significant role in the high incidence of osteoporosis and periodontosis later in life. This can be intensified in the treatment of life-threatening hypercalcemia of malignant disease and hyperparathyroidism.
The use, not only of phosphate therapy (which has a distinct risk of metastatic calcinosis), but of high-dosage calcemic agents during infancy (to counter hypocalcemia and refractory osteopenias), and to treat postmenopausal, senile, or disuse osteoporosis, also intensifies magnesium loss and metastatic calcification. Such treatment also increases bone mineralization, but in the absence of optimal magnesium, the bone has abnormal matrix. Such treatment is likely to cause increased bone density, but decreased bone elasticity, with resultant marblelike, brittle bones. Renal dysfunctions-tubular acidosis, aminoaciduria, and calcinosis and calculi- might also result from magnesium deficiency, intensified by calcemic therapy.
This book presents evidence that early investigation of the magnesium status is important. Whether use of magnesium supplements during gestation and infancy will reduce the incidence of some of the indicated congenital anomalies will require many years to ascertain. Clues might be obtained from experimental models, prepared so as to mimic some of the nutritional imbalances, and to exaggerate magnesium deficiency, such as might be found with genetic magnesium malabsorption or renal wastage. Since magnesium administration is benign (unless there is renal failure), it is proposed that prophylactic and therapeutic trials are justifiable.
This particularly true for patients with premature cardiovascular disease, or for subjects with familial histories suggesting high risk of early ischemic heart disease or strokes. It is also true for patients with the bone disorders cited, and for those with functional renal disorders such as tubular acidosis and aminoaciduria, and for patients with calcific urinary tract disease.
There are serious problems in assessing the magnesium status of patients, probably the most important reason that, despite the ubiquity of this element and its importance in so many enzyme systems and in function and structure of vital organs and bones, magnesium is usually one of the last clinical parameters to be explored (Whang et al., 1976/1980). When levels are sought, the results are often misleading. Each means of evaluation has its limitations, and in order to determine whether a patient is magnesium deficient (unless the deficiency is so profound as to cause unquestioned hypomagnesemia), a combination of approaches may be necessary. First of all, although magnesium is an intracellular cation, second in concentration only to potassium [the retention of which is dependent on magnesium-dependent enzymes (Reviews: Wacker and Vallee, 1958; Whang et al.1967; Whang, 1968, 1971; Seelig, 1972; Whang and Aikawa, 1977)], serum magnesium is generally the only parameter explored. Unfortunately, the reliability of serum magnesium values is dubious as an index of body levels, and even as an indication of abnormal blood levels, particularly when wide ranges of serum or plasma magnesium levels are accepted as "within normal limits" (infra vide).
With the limitations of serum magnesium values, the clinician must rely on indirect tests of magnesium metabolism, determinations of cellular magnesium levels (e.g., blood cells, skeletal muscle) generally being unattainable and not standardized (infra vide). Metabolic balance studies have provided important baseline data regarding magnesium requirements of normal subjects (Seelig, 1964). However, metabolic research units are necessary to obtain reliable results and the procedure is time consuming and cumbersome. Furthermore, when used with patients who have intrinsic (isolated, possibly familial) magnesium malabsorption, or who have renal magnesium wastage as a result of renal disease or a genetic trait, the results can be misleading. Prolonged studies, and periods of magnesium restriction (which might be of risk to patients with underlying magnesium deficiency) would be necessary to separate those who do not retain magnesium because their tissue stores are ample from those who have a metabolic abnormality resulting in magnesium malabsorption or renal wastage. Also, such studies are inapplicable to patients who require medication that can interfere with the intestinal absorption or renal tubular reabsorption of magnesium.
At this time, the most reliable method of evaluating a patient's magnesium status is determination of its 24-hour urinary output before and after a parenteral magnesium load, and evaluating the percentage retention in terms of renal function and serum magnesium levels (infra vide).
A.1.1. What is the Normal Range?
Serum magnesium levels are normally maintained within a very narrow range, with a coefficient of variation of only 10% to 20% (Alcock et al., 1960; Hanna, 1961b; Prasad et al., 1961; Stewart et al., 1963; Ginn, 1968; Hunt, 1969; Henrotte and Durlach, 1971; Rousselet and Durlach, 1971; Seelig and Berger, 1974), unless there is a profound deficiency, or magnesium load in the face of renal failure. Thus, the serum or plasma magnesium level is not a reliable index of magnesium deficiency (Walser, 1967; Gitelman and Welt, 1969; Henrotte and Durlach 1971; Rousselet and Durlach, 1971). To make matters worse, there are many sources of error even in the most reliable technic available, atomic absorption spectrophotometry (Table A-1). Thus, each laboratory should establish its own mean and narrow range of normal values (Hunt, 1969; Seelig and Berger, 1974). Not acceptable as "within normal limits" are values that fall between 1.5 and 2.5 mEq/liter, a wide range obtained from data reported from many laboratories, and that has been designated as the normal "reference" range (Unsigned, N Engl J M 1974) (Table A-2A) and (Table A-2B).
{kind=link}
{kind=link}
{kind=link}
Rarely are efforts made to differentiate among ionized, complexed, or protein-bound fractions of serum magnesium, since expensive equipment is required for measurement of the protein-bound and diffusible fractions (Silverman et al. 1954; Prasad et al. 1961; Walser, 1967; S. P. Nielsen, 1969; Cummings et al., 1968; Voskian et al., 1973). There are no readily available means of measuring ionized magnesium. Because many factors influence the degree of binding, complexing, or chelating of magnesium, the total content of magnesium in the serum is not simply related to the availability of magnesium, either extra- or intracellularly. For example, experimental dietary magnesium deficiency has caused an increase in the protein-bound fraction (Hoobler et al.., 1937; Morris and O'Dell, 1969) or decreased total and ultrafiltrable fraction (Woodward and Reed, 1969). Clinical magnesium deficiency of intestinal malabsorption (Silverman and Gardner, 1954) and of hepatic cirrhosis (Prasadet al., 1961) is associated with decreased protein-bound magnesium, and increased ultrafiltrable fraction. Possibly, the thyroid hormone affects the degree of protein-binding of magnesium (Soffer et al., 1941; Dine and Lavietes, 1942; Silverman et al., 1954; Prasad et al., 1961), but there is no accord as to the degree or the mechanism. The level of plasma citrate, which complexes part of the ultrafiltrable fraction of magnesium, is influenced by growth hormone (Hanna et al., 1961), adrenocorticosteroid hormone (Walser et al., 1963), estrogen (N. F. Goldsmith et al., 1970) and vitamin D (Carlsson and Hollunger, 1954). Not all of the magnesium-complexing or chelating anions in the body are known. Magnesium complexes comprise 14% of the total plasma magnesium: Mg citrate, 4%; magnesium HPO4 3%; unidentified complexes, 6% (Walser, 1961).
Furthermore, even the way the blood is drawn can affect the serum or plasma magnesium values. Levels are lower in serum from blood obtained quickly after applying the tourniquet than after prolonged venous stasis (Whang and Wagner, 1964, 1966; S. P. Nielsen, 1969). This may be referable to the egress of cellular magnesium in hypoxic states (Engel and Elin, 1970; Hochrein, 1966; Hochrein et al., 1967). In addition, dehydration or acidosis can yield spuriously high serum magnesium levels.
Until it is feasible to demonstrate cellular magnesium deficiency in a tissue that has metabolic characteristics and magnesium exchangeability, similar to that of the metabolically active tissues, conclusions as to the importance of magnesium in physiologic processes will remain open to dispute. Enzymatic studies of magnesium-dependent enzyme systems are important in providing clues as to the effects of suboptimal magnesium concentrations in the body, cells, and cell-fractions (Reviews: Wacker and Vallee, 1964; Walser, 1967; Wacker and Parisi, 1968; Heaton, 1978). Direct determinations of cellular magnesium levels, however, are necessary for clinical evaluation of the changing magnesium status of individuals under the influence of diseases and treatment regimens that alter magnesium retention.
A.2.1. Erythrocyte Magnesium
Tissue magnesium levels have most frequently been estimated on the basis of analysis of erythrocytes for magnesium. Despite investigations for over 40 years, erythrocyte magnesium levels have not proven a reliable source of information as to the clinical magnesium status. Analyses of findings from over 20 studies indicate that the means of RBC-Mg are between 1.9-3.1 mmol/liter (1.8-6.2 mEq/liter) (Review: Henrotte and Durlach, 1971). The ranges, given as normal in individual studies, are often even wider (Table A-3). Such broad "normal" ranges make it difficult to detect significant changes in abnormal conditions.
{kind=link}
Many procedures have been utilized in the effort to minimize sources of error, and to obtain uniformity of results. The first controlled study (Greenberg et al., 1933) showed that direct measurement of the magnesium content of saline-washed erythrocytes, and indirect measurement (subtracting plasma magnesium from whole blood magnesium, and correcting for differences in hematocrits) yielded comparable results. Washing erythrocytes with isotonic saline (Greenberg et al., 1933) or with buffer (Valberg et al., 1965) did not cause loss of cellular magnesium. Attempts to improve validity of magnesium analysis of packed erythrocytes have included: (1) correction for trapped plasma and for differences in hemoglobin (Valberg et al., 1965; S. Hellerstein et al., 1970); (2) use of cation-exchange resins to remove all Mg from cell fragments and hemolystates (Hunt and Manery, 1970; Frazer et al., 1972), (3) rapid separation cells and protein precipitates to prevent elution of magnesium into the hemolysate (Stephan and Speich, 1972; Welin and Speich, 1973); (4) saponification of unwashed erythrocytes without deproteinization (Rousselet and Durlach, 1971); (5) measurement of magnesium in washed and ashed cells, in terms of mg/g cells (Paschen et al., 1971), and (6) of weight per cell count (Valberg 1965; Rosner and Gorfein, 1968).
Except for those who measure magnesium in ashed erythrocytes, and those using cation exchange resin on cell ghosts and hemolysates (Hunt and Manery, 1970; Paschen et al., 1971), the levels are determined in hemolysates, the cell ghosts being discarded with the rest of the precipitated protein. Most of the erythrocyte magnesium is in the hemoglobin, in association with the organic phosphates and enzymes, and is released when the cells are disrupted (Rose, 1968; Bunn et al., 1971). A portion of the erythrocyte magnesium, however, is bound to the membranes (Carvalho et al., 1963), and remains even after repeated washing (Fujii et al., 1973; Sato and Fujii, 1974). Although only 2-6% of the total erythrocyte magnesium is present in the membranes, which are separated from washed erythrocytes (Fujii et al., 1973), the hemolysis procedure may provide a source of error, particularly if the disrupted cell membranes remain in contact with the hemolysate for different lengths of time. Divalent cations (Mg2+, Ca2+ in the suspending medium are readily bound to the disrupted membranes, both internal and external surfaces of which are exposed to the medium (Sato and Fujii, 1974). Large and variable amounts of the cations are taken up by the stroma.
The significantly higher magnesium levels in reticulocytes and young erythrocytes than in old erythrocytes (Henriques and Orskov, 1939; Bang and Orskov, 1939; Dahl, 1950; Ginsberg et al., 1962; R. Bernstein, 1959) are probably responsible for the major discrepancies in reported normal erythrocyte levels. In experimentally induced reticulocytosis, the erythrocyte magnesium was 28.4 mEq/liter (in rabbits with 85% reticulocytes), in contrast to 7.8 mEq/liter in control rabbit erythrocyte (Ginsberg et al., 1962). Patients with high reticulocyte counts have higher erythrocyte magnesium levels than do those with low reticulocyte counts (Dahl, 1950; Ginsberg et al.1962: R. Bernstein, 1959). For example, a patient with 89.7% reticulocytes had RBC Mg of 4.0-4.7 mEq/liter (Ginsberget al., 1962). Because reticulocytes and young erythrocytes remain at the top of the centrifuged column of red cells, and the older erythrocytes sediment to the bottom (Keitel, 1955; R. Bernstein, 1959; Ginsberg et al.1962), when erythrocytes are analyzed for magnesium, either the entire column should be studied (S. Hellerstein et al., 1960) to minimize the risk of obtaining aliquots from different levels, or only the lowest level, where the old erythrocytes are found, should be analyzed (Ross et al.1976/1980).
It is possible that marginal abnormalities in magnesium levels might be masked by procedures that measure only the hemolysate magnesium, even when the membranes are immediately separated from the hemolysate. In a study of RBC magnesium of anemic children (S. Hellerstein et al., 1970), their erythrocytes had significantly higher-than-control magnesium, in terms of Mg/cell solids, but not in terms of Mg of cells or of cell water. In a study of erythrocytes from convalescent cardiac patients (Borun, 1963) the older erythrocytes (from the botton of the centrifuged column of cells) had higher magnesium levels than did the young erythrocytes, in terms of Mg/L cell water, but not by dry weight. Whether use of agents that cause magnesium loss contribute to decreased magnesium levels in reticulocytes formed [e.g., during magnesium loss of active treatment of congestive heart failure (Wacker and Vallee, 1958; Seller et al., 1966; Wacker and Parisi, 1968; Seelig, 1972; Lim and Jacob, 1972b)] seems plausible. There is direct evidence that erythrocyte-magnesium levels are significantly below normal in patients with congestive heart failure (Seller et al., 1966; Lim and Jacob, 1972b).
The erythrocyte membrane instability (and tendency toward hemolysis) that is found in magnesium deficiency (Larvor et al., 1965; Erlandson and Wehman, 1966; LaCelle and Weed, 1969; Cohlan et al., 1970; Oken et al., 1971; Battifora, 1971; Elin et al. l971b; Elin, 1973; Piomelli et al., 1973; Elin, 1976/1980) may provide another source of error when erythrocyte magnesium is obtained by analysis of the hemolysate. With progressive magnesium deficiency, associated with low intra- and extracellular magnesium levels, there is increased fragility of the erythrocytes and shortening of survival time (Elin, 1973). One may speculate that the defective erythrocyte membranes may have defective binding of magnesium, thereby releasing more during laboratory-hemolysis. Whether this might yield spuriously higher erythrocyte magnesium values in hemolysates, thereby masking cellular deficiency, remains to be investigated.
Still another limitation of erythrocyte magnesium is its unreliability, as an index of magnesium status at the time of the analysis. There is poor correlation between plasma and erythrocyte levels (Wallachet al., 1962; Valberg et al.1965; Hellerstein et al., 1970; Ross et al., 1976/1980), although in chronic conditions, such as long-term magnesium deficiency, malnutrition, chronic liver disease, and hypothyroidism, there may be low plasma and erythrocyte magnesium levels. Acute two- to fourfold increases in plasma magnesium (by intravenous infusions) are not accompanied by changes in erythrocyte levels (Wallach et al. 1962). Hemodialysis of hypermagnesemic patients, with magnesium-free or -low dialysates, has had little or no effect on erythrocyte magnesium (Paschen et al., 1971). Under usual circum stances, the red cell membrane permits only slow diffusion of magnesium (Wallach et al. 1962; Ginsberg et al. 1962). This is reflected by the very slow uptake of28Mg by erythrocytes (Zumoff et al. 1958; Care et al. 1959; Aikawa et al. 1960c; Rogers, 1961; Ginsberg et al. 1962; Aikawa, 1965; Hilmy and Somjen, 1968).
It has long been recognized that erythrocyte magnesium levels do not fall as quickly or as much as does plasma magnesium in acute magnesium deficiency (Tufts and Greenberg, 1937). However, lesser degrees of magnesium deficiency, if prolonged, can cause profound drops in erythrocyte magnesium, to half control values (Elin et al., 1971a,b). Most data indicate that erythrocyte magnesium levels reflect: (1) the magnesium status at the time of erythropoiesis (Tufts and Greenberg, 1937; MacIntyre et al. 1961; Dunn and Walser, 1966; Walser, 1967; Hellerstein et al., 1970; Elin et al. 1971a,b) and the age of the red cells (Henriques and Orskov, 1939; Bang and Orskov, 1939; Dahl, 1950; Bernstein, 1959; Ginsberg et al., 1962). Thus, the erythrocyte magnesium level is dependent, more on the age mix of the cells in the sample studied, than on the magnesium status at the time it is taken.
A.2.2. Skeletal Muscle Magnesium
Analyses of skeletal muscle biopsies has been recommended as a more useful clinical index of intracellular magnesium than erythrocyte or plasma magnesium (MacIntyre et al, 1961; Dunn and Walser, 1966; Seller et al. 1966; Walser, 1967; Lim et al., 1969a,b; Drenick et al., 1969; Lim and Jacob, 1972a,b). The values for normal human skeletal muscle magnesium are similar to those for normal myocardium (Lim et al., 1969b; Bertrand, 1967; Tipton and Cook, 1963). (For tabulation of data from cattle, horses, pigs, dogs, and rodents, see Walser, 1967.) However, the muscle magnesium levels in experimental animal and human magnesium deficiencies and in clinical magnesium deficiency do not always indicate loss of magnesium. In many studies of acute magnesium deficiency, muscle levels remained essentially unchanged, or decreased only slightly (Cunningham, 1936a; Watchorn and McCance, 1937; Cotlove et al., 1951; Blaxter and Brook, 1954; Morris and O'Dell, 1961, Ko et al. 1962; Welt, 1964; Dunn and Walser, 1966; Bradbury et al. 1968; Woodward and Reed, 1969; Elin et al. 1971).
Because skeletal and cardiac muscle are structurally more comparable than the myocardium is to other tissues, and the magnesium levels in the two tissues are similar (Tipton and Cook, 1963; Wallach et al., 1966a,b, 1967; Lazzara et al., 1963; Walser, 1967), muscle biopsies would seem to provide a useful index of myocardial magnesium levels. However, the exchangeability of skeletal muscle magnesium is much slower than is that of more metabolically active tissues, such as the heart, kidneys, and liver (Aikawa, 1963; Gilbert, 1960; Review: Walser, 1967) (Table A-4). Furthermore, young animals show lower levels of muscle magnesium more rapidly when magnesium deficient than do older animals (Tufts and Greenberg 1937b; Cotlove et al. 1951; MacIntyre and Davidsson, 1958; Morris and O'Dell, 1961; Smith et al., 1962) and greater losses are seen in chronic than in acute deficiencies (MacIntyre et al. 1961; Montgomery 1960, 1961a; Booth et al. 1963; Whang and Welt, 1963). It is possible that the failure of skeletal muscle magnesium to show a significant response to acute deficiency might reflect a high percentage of tightly bound magnesium in skeletal magnesium (Elin et al., 1971)
{kind=link}
Since cardiovascular and renal tissues are vulnerable to damage caused by acute and chronic magnesium deficiencies, it is important to select a tissue for analysis that is more likely than are plasma, red cells, or muscle to reflect the magnesium status of those organs.
It seems likely that leukocytes, which are the most readily available nucleated, metabolically active cells, should provide a more reliable index of magnesium levels of such tissues as the heart and kidneys than the serum, erythrocytes, or skeletal muscle. Lymph nodes and spleen, for example, have magnesium exchangeability, in terms of speed of uptake of the isotope and the concentration attained by 24hours, closest to that of heart, kidneys, and liver. We are attempting to develop a procedure for isolating white blood cells, by a means that does not traumatize the membranes, and that will be adaptable to laboratories lacking sophisticated equipment (Ross et al. 1976/1980). In our first venture, we analyzed the total white cell isolate, using a modification of the Boyum (1968) procedure (dextran/hypaque sedimentation), without attempting to separate the small and large mononuclear cells from the polymorphonuclear cells. We have now simplified, somewhat, a time-consuming, cumbersome procedure that has the intrinsic defect of analyzing mixed cells, and are studying lymphocyte magnesium levels (Ross, Seelig, and Berger, in preparation). A similar method has been used by M. P. Ryan and his colleagues in monitoring lymphocyte magnesium levels in patients with congestive heart failure (Counihan et al. 1978a,b). Since there is no standard procedure, nor standard values for white blood cell magnesium, listing our values would be premature until considerably more data have been accrued.
The most practical means of evaluating the magnesium status relatively quickly, and with facilities that are readily available, is the determination of 24-hour urinary magnesium output before and after a magnesium load. (We have already discussed possible risk of magnesium loading in the presence of hypercalcemia. Renal failure also militates against a test that might result in marked hypermagnesemia.)
Fitzgerald and Fourman (1956) found that two volunteers, who retained almost none of injected magnesium, as the sulfate, during a control period of adequate magnesium intake, retained 25% and 42% of parenterally administered magnesium (49 and 82 mEq over 2 and 3 days, respectively). Patients whose chronic magnesium deficiency secondary to steatorrhea might have been missed on the basis of their serum magnesium levels (which were 1.39, 1.67, 1.69, and 1.75 mEq/liter), retained 37% to 79% the first 24 hours after receiving 84 mEq of magnesium, given as magnesium sulfate or chloride infusions (Fourman and Morgan, 1962). Thoren (1963) then confirmed earlier observations that normal subjects excrete at least 80-85% of parenterally (i.v. or i.m.) administered magnesium within 24 hours, and found that many of his surgical patients retained considerably more. He concluded that patients who retain more than 20-25% of magnesium (e.g., 20 mEq in two divided doses) are probably repleting a deficit. He commented, however, that patients with magnesium deficiency due to renal magnesium loss, might not be detected by this test. Jones and Fourman (1966) extended the studies of percentage retention of parenteral magnesium infusions (84 mEq) to patients with hypoparathyroidism and found that all seven retained more than 50%, three retaining about 80%.
Application of the magnesium-loading test for proof of suspected magnesium deficiency in infancy was first reported in England (Wilkinson and Harris, 1969; Harris and Wilkinson, 1971). These investigators, who had found magnesium therapy useful over a ten-year period, in the treatment of infants in poor condition because of persistent diarrhea, other causes of loss of gastrointestinal fluids, or who were unresponsive to calcium or other therapy, reported that they were able to prove magnesium depletion in 20 of 29 cases in which the magnesium-loading dose (0.5 mEq/kg) was used, 40% or more of the dose being retained. They administered between 0.24 to 5.71 mEq Mg by mouth in 4, by gastrostomy or nasogastric tube in 2, intramuscularly in 1, and intravenously in 22. Among 9 whose serum magnesium had been measured, it was above the normal range of 1.4 to 1.9 mEq/liter in 2, one of whom retained over half of the test done. The serum magnesium was normal in 4 patients, 3 of whom were magnesium deficient. All 3 with hypomagnesemia retained over 70% of the test dose. Caddell et al. (1973b) and Caddell and Olson (1973) similarly found that the lowest magnesium plasma levels (in 40 babies with kwashiorkor or marasmus or both) were correlated with the highest magnesium retentions, and that some with normal plasma magnesium levels had high retentions. These investigators did not find low preload magnesium urinary excretion to be a helpful guide; 7 of 25 who excreted less than 1 mEq of magnesium per 24 hours retained a mean of on 23.3% of the magnesium load and clinical magnesium deficiency was not diagnosed. Caddell (1975) and her colleagues (Caddell and Olson, 1973; Caddell et al. 1973b, 1975a; Byrne and Caddell, 1975) have evaluated magnesium-load test in neonatal, normal, and low-birth-weight infants and infants during the first few months of life, and designed a shorter test (infra vide), as well as using this test to evaluate the magnesium status postpartum (Caddell et al. 1973a, 1975b). In the magnesium-loading studies of postpartum women, Caddell et al, (1973, 1976) found that among Thai women with ample magnesium intakes, the postpartum women retained more magnesium than did nulliparous young women, but not nearly as much as did many of the American women (particularly young multiparous women) However, except for women with plasma magnesium levels below 1.2 mEq/liter, the amount of magnesium retained was not reflected by the plasma levels.
Although, ideally, it is desirable to obtain a 24-hour urine sample for base-line magnesium levels (and for creatinine output to permit evaluation of renal function before and after the magnesium load), the clinical status may be too precarious to permit so long a delay before instituting magnesium therapy when there are signs suggesting its depletion. In that event, a single pretreatment urine and blood sample for magnesium and creatinine levels must suffice, and the 24-hour posttreatment urine collected for analysis. Those, whose test is part of a diagnostic procedure, should have magnesium laxatives and antacid withheld for 48 hours before the pretreatment collection and during the test. If medically acceptable, withhold strong magnesium-wasting diuretics, such as furosemide or ethacrynic acid, or substitute a thiazide diuretic for the duration of the test.
A.3.1.1. Adults: Intramuscular Load
After collecting urine for 24 hours and taking a blood sample for magnesium and creatinine levels, 2 ml of 50% MgSO4 (100 mg of magnesium) should be injected deep into each buttock. Collect the next 24-hour urine, and draw blood at the end of the collection period for magnesium and creatinine analysis. It is often of value to have the specimens analyzed for additional electrolytes, such as calcium, sodium, potassium, and phosphorus. Subtract the amount of magnesium in the preload 24-hour urine from that in the postload 24-hour urine and calculate the percentage of the load that was retained.
A.3.1.2. Adults: Intravenous Load
The procedure is as above, except that the magnesium load (0.4 to 0.5 mEq/kg, as magnesium sulfate or magnesium chloride, diluted in 100 cc 6% dextrose in water or 0.9% saline) is given over a 45-minute period.
A.3.1.3. Infants: Intravenous Load
The procedure is as for adults, with the time for delivery extended to 1-1 1/2 hours. Harris and Wilkinson (1971) caution that no talcum powder should be used during collection periods, and that the collecting vessel (i.e., plastic bag) should be rinsed at least six times with de-ionized water.
A.3.1.4. Infants: Intramuscular Load
Caddell et al, (1975) and her co-workers (Byrne and Caddell, 1975; Caddell et al., 1975a) have modified the test to allow for shorter collection periods for infants up to 6 months of age. Preload plasma cations and 8-hour urinary levels of magnesium, calcium, potassium, sodium, and creatinine are determined. An intramuscular injection of 50% sulfate (0.12 ml/kg, equivalent to 0.49 mEq/kg of body weight) is given to infants whose plasma magnesium levels do not exceed 2.0 mEq/liter, who are well hydrated, and who have good renal function. Caddell cautions that although even premature neonates can excrete an excessive magnesium load (as has been shown by the rapid drops of serum magnesium levels in symptomatic hypermagnesemic infants born to eclamptic mothers given high doses of magnesium within 24 hours of delivery) (Brady and Williams, 1967; Soka et al., 1972), neonatal infants have immature renal function (Rubin et al. 1949; Wilkinson, 1973). Thus, the risk of producing hypermagnesemia and of reciprocally increasing urinary calcium excretion must be kept in mind. Caddell (1975) has observed that most of the magnesium load was usually excreted during the first 8 hours postload, although in a few instances neonatals excreted more magnesium in the second 8-hour period. The 24-hour reading usually provided a reliable reading of the amount retained. Infants who retained more magnesium had lower plasma levels of magnesium than did those who retained less. For example, full-term infants who retained 80% of the load had preload plasma magnesium levels of 1.50 mEq/liter; the prematures with plasma magnesium levels averaging 1.59 mEq/liter retained 85.67 ± 2.2 of the load. Those with preload plasma levels of 1.77 and 1.90 mEq/liter (full term and premature) retained 28.2 ± 3.04 and 21.5 ± 0.89, respectively (Byrne and Caddell, 1975). However, despite the grouped evidence correlating low plasma magnesium levels with high retentions, the authors pointed out that in their series individual instances of magnesium deficiency of infancy would have been infrequently diagnosed on the basis of the plasma values alone. The infants had higher plasma magnesium levels at the end of the load test than at the beginning, an effect attributed to normalization of low initial values and incomplete renal clearance of the load (Caddell, 1975).
Freeman and Pearson (1966) reported a patient with renal magnesium wastage, detected because the amount of (preload) magnesium excreted was inappropriately high in view of her hypomagnesemia, and who exhibited only partial renal conservation of magnesium on moderate reduction of her magnesium intake. They pointed out that a prerequisite for the magnesium-loading test is normal renal mechanisms for conserving magnesium.
Parfitt (1976/1980) has developed a model for assessing the tubular reabsorption of magnesium that plots data for UMgV/GFR against the plasma magnesium level. He points out that short-term renal conservation of magnesium passively reflects the fall of plasma magnesium levels. Long-term depletion results in active renal magnesium conservation, which might result from an increase in its maximum tubular (Tm) reabsorption. However, in a prolonged magnesium depletion study (Shils, 1969a), no increase in tubular magnesium reabsorption was observed, and a maximum Tm has, in fact, been demonstrated for magnesium (Barker et al., 1959; Averill and Heaton, 1966; Massry et al., 1969). Thus, agents that cause renal magnesium wastage do so by lowering TmMg/GFR transiently or permanently. In the case of the metabolic abnormality that interferes with renal tubular magnesium reabsorption, the TmMg/GFR is abnormally low.
===========================================
No comments:
Post a Comment