

MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF DISEASE
Early Roots of Cardiovascular, Skeletal
and Renal Abnormalities
and Renal Abnormalities
Goldwater Memorial Hospital
New York University Medical Center
New York, New York
1980
New York University Medical Center
New York, New York
1980
Part II: Chapter 5
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
Cardiovascular diseases continue to represent the major cause of morbidity and mortality in the developed countries, especially in young men, despite the considerable efforts and funds expended in the effort to explain and reverse the atherosclerotic process by studying and modifying fat intakes. Ischemic heart disease (IHD) is responsible for over 54% of all deaths in the United States (U.S. Dept. HEW, 1970); it is the major cause of death in most affluent communities (Editorial, Brit Med J, 1972). That the problem has increased during this century, particularly in young and middle-aged men, is indicated by two types of studies: (1) retrospective analyses of large numbers of necropsies (over 6000 each) in a large city hospital (Saphir et al., 1956) and from the Armed Forces Institute of Pathology (Pettyjohn and McMeekin, 1975); and (2) examination of the hearts of military men coming to autopsy in World War II (Yater et al., 1948, 1951; Moritz and Zamcheck, 1947), the Korean War (Enos et al., 1955), and the Vietnamese War (Macomber, 1971; McNamara et al., 1971; Wroblewski, 1971), and of victims of aircraft fatalities (Glantz and Stembridge, 1959). The study at Michael Reese Hospital in Chicago (Saphir et al., 1956) showed that there was an increase in frequency of coronary artery disease in subjects under 50 from 5.9% in 1920-1939, to 14.1% in 1940-1949, to 25.5% in 1950-1953. This did not take into account the almost twofold greater frequency of IHD in men than women. Pettyjohn and McMeekin (1975) found that 13% (816) of 6500 autopsied cases from aircraft accidents had been diagnosed as having had preexisting heart disease. Of those 816 cases (592 military and 135 civilian), 89.1% had coronary artery disease. Among the 380 men, 20-34 years of age, an upward trend was noted in the incidence of moderate to severe coronary artery disease from 1960-1964 to 1965-1969. (Too few autopsies were available for the 1970-1974 study for valid comparison.) In the studies of soldiers in the last three wars, startlingly high numbers of young men were found to have IHD. Yater et al. (1948) studied heart tissue from 866 American World War II soldiers, between 18 and 39 years of age, who developed IHD; 450 of these were examined at autopsy. From the incidence among soldiers, they estimated that the IHD death rate, per 100,000 men was less than 0.1 at 18-19, 1.0 at age 25-29, 3.4 at age 30-34, and 12.7 at age 35-39. Moritz and Zamcheck (1946) reported 115 sudden deaths from IHD in additional young soldiers. In the study of 300 American soldiers killed in Korea (Enos et al., 1955), 77% had histologic evidence of coronary disease; the average age was 22.1 years. The 1959 study of material from Air Force fatalities (Glantz and Stembridge, 1959) showed that 70% of 222 men of 20-44 had coronary disease, the highest incidence being in men 30-34, 35-39, and 40-44 years of age who had moderate to advanced arteriosclerosis in 32%, 26%, and 50%, respectively. There was a difference of opinion as to whether there was, indeed, a lower incidence of coronary artery disease among American soldiers killed in Vietnam (McNamara et al., 1971; Macomber, 1971; Wroblewski, 1971). Pettyjohn and McMeekin (1975) analyzed the factors contributing to the seeming decline in IHD incidence (McNamara et al., 1971) and attributed this finding to a difference in parameters used in classification of disease. The relative increase in mortality rates from IHD in the younger age groups has been confirmed by the international studies (International Workshop in Cardiovascular Disease, 1959-1969; Fejfar, 1974) even from groups employing measures to lower blood cholesterol levels (Fejfar, 1974).
As a result of the failure to prove that the incidence of deaths from IHD can be lowered by changing the fat intake of patients with the disease (Editorial, Brit Med J, 1972, 1976b; Stolley, 1972; Fredrickson, 1972; Fejfar, 1974), there has been revival of interest in the likelihood that adult cardiovascular disease has its roots in infancy, and that that is the time to change the fat in the diet (U.S. Dept. HEW, 1970; Glueck and Tsang, 1972; Glueck et al., 1972; 1974a). This approach is based on three findings: (1) the detection of fatty intimal streaks in arteries of infants and children (Duff and McMillan, 1951; R. Holman et al., 1958; R. Holman, 1961; Reisman, 1965; Strong and McGill, 1969); (2) the correlation of hyperlipidemia with increased risk of early arteriosclerosis (Gofman et al., 1950; Keys, 1956; Gertler et al., 1959; Berenson et al., 1974); and (3) the evidence that children of victims of early heart attacks often have hyperlipidemia (Tamir et al., 1972: Glueck et al., 1974b; H. Chase et al., 1974). Furthermore, large-scale screening programs have shown that three-year-old children already have cholesterol levels similar to those of young adults (Berenson et al., 1974). At present, it is considered feasible only to screen children with parenteral histories of early IHD (H. Chase et al., 1974; North, 1975; Laird, 1975). A general change of diet, so as to institute hypolipidemic regimens has been suggested (U.S. Dept. HEW, 1970). Altering the fat content of infants' diets substantially has been criticized because not all of the etiologic factors in arteriosclerosis are known, and because the results of the field trials have not yet proven that substituting unsaturated for saturated fatty acids will prevent coronary heart disease, even though they have lowered blood lipids (Stolley, 1972; Frederickson, 1972; C. Lowe, 1972; Levy et al., 1974; North, 1975; Laird, 1975). Furthermore, the potential risks of such diets remain to be ascertained (Foman, 1974; Schubert, 1973; Laird, 1975; Glueck et al., 1975/1977.
Thus, the need for searching out coronary-risk indicators persists (Blackburn, 1974). Changes in the musculoelastic layer of coronary arteries of infants and children are again being considered as the possible initial lesions in the atherosclerotic process (Neufeld, 1974; Danilevicus, 1974). The first visible changes in the internal elastic membrane, its splitting or fragmentation, are seen within a few days after birth (or in some cases in stillborn infants) and become more prominent in the first month of life (Bertelsen and Jensen, 1960; Bertelsen, 1961; Neufeld and Vlodaver, 1968, 1971, 1974; Neufeld, 1974). These are changes that have long been proposed as the first departure from normal, and that should be considered a manifestation of early arteriosclerosis and the basis for development of atherosclerotic lesions (Merkel, 1903; Jores, 1924; Minkowski, 1947; Fangman and Hellwig, 1947; Levene, 1956; Moon, 1957; Pizzagalli and Bertana, 1959; Bertelsen, 1961; Kaunitz, 1961; Gillot, 1962).
Many factors contribute to the metabolic abnormalities that lead to different blood, arterial, and cardiac biochemical, functional, and histological changes that represent aspects of the complex of cardiovascular diseases. Vitamins B6 and E have been suggested as protective against arteriosclerosis. Vitamin B6 has been suggested by Rinehart and Greenberg (1949, 1951, 1956), Moon and Rinehart (1952), Moon (1957, 1959), Boxer et al. (1957), Hass (1961), and Levene and Murray (1977). Vitamin E has been suggested in peripheral disease, e.g., intermittent claudication, by Livingstone and Jones (1958), Haeger (1968, 1973), Larsson and Haeger (1968), and Williams et al.(1971); in thrombotic disease by Zierler et al. (1948), Ochsner (1951), Suffel (1956), and Kawahara (1959); and in the controversial use in heart disease by Vogelsang et al. (1947; publications of the Shute Institute). Pyridoxine/blood- and tissue-lipid interrelationships have long been known (Birch, 1938; Medes and Keller, 1948; Schroeder, 1955; Shah et al., 1960; G. Emerson et al., 1960; Lupien, 1968), and combinations of the vitamins, sometimes with A (Hammerl and Pichler, 1960) proposed. Vitamin C has been shown to lower plasma cholesterol levels, and by inference atherosclerosis (Spittle, 1970, 1971; Anderson et al., 1972). However, hypercholesterolemia has been produced in rats by supplements of vitamin C equivalent to excesses of less than one gram over that in the diet, an effect attributed to ascorbic acid induced production of high zinc/copper ratios (Klevay, 1977). Thiamine deficiency has been shown to increase the hepatic synthesis of lipids by rats; hypertriglyceridemia of magnesium deficiency develops in the presence of adequate or excess thiamine, but not in double deficiency (Itokawa et at., 1973). Excess vitamin D increases arteriosclerosis both in experimental animals and in man-infants, children, and adults. In his epidemiologic correlation of only slightly higher than recommended intakes of vitamin D with increased incidence of myocardial infarction, Linden (1974b) suggested that the hypocholesterolemic effect of vitamin A might protect against the hypercholesterolemic action of vitamin D. Additional studies confirm that experimental A deficiency increases both atherosclerosis and cholesterol blood levels (Bayer et al., 1972; Bonner et al., 1973). In 1962, I. Clark and Bassett showed that vitamin A decreased other manifestations of vitamin D toxicity: osteolysis and renal and arterial calcinosis. As early as 1939, Reed et al. reviewed the data on vitamin D toxicity and reported that in the absence of vitamin A, the lesions of hypervitaminosis were worse.
Several of these vitamins are of interest in this presentation because they affect magnesium metabolism, influence the response to magnesium, or affect magnesium requirements. For example, vitamin B6 deficiency causes arterial lesions (Rinehart and Greenberg, 1949, 1951) very much like those of magnesium deficiency (Hass, 1961), and gestational B6 deficiency has been blamed for the elastica damage of neonatal arteriosclerosis (Levene and Murray, 1977), in which magnesium deficiency is implicated in this book. Interrelationships of magnesium and pyridoxine metabolism have been reviewed by Durlach (1969b). The response of B1-deficient animals (Zieve et al., 1968a,b; Zieve, 1969) and man (Zieve, 1975) is magnesium dependent. The cardiovascular lesions of vitamin D excess might be partially implemented by magnesium loss. Vitamin D increases magnesium loss by increasing its renal excretion relative to its absorption (Hanna, l961b; Richardson and Welt, 1965; Wallach et al., 1966). Infants and children with hypervitaminosis D, who develop hypercalcemia, the supravalvular aortic stenosis syndrome (SASS), and other stenotic and hypoplastic lesions of the greater arteries, also develop peripheral arterial lesions (including atheromata and calcinosis), hypercholesterolemia, and hypertension. Since vitamin D excess causes calcium retention as well as magnesium loss and high calcium/magnesium plasma ratios have produced increased arterial resistance (Review: Haddy and Seelig, 1976/1980, and infra vide), the combination of low magnesium stores at birth and high vitamin D and calcium intakes in infancy can be responsible for several metabolic and histologic aberrations leading to cardiovascular diseases.
The similarity of the arterial lesions, and of the microfocal myocardial necrosis seen in the infants, to those produced experimentally by essentially "pure" magnesium deficiency (Seelig and Haddy, 1976/1980) has suggested magnesium deficiency during gestation and infancy. Furthermore, magnesium deficiency, even in the absence of hypervitaminosis D, has been shown to cause abnormal changes in blood lipids (Review: Seelig and Vitale, 1971/1973).
Epidemiologic data point both toward magnesium as a protective factor against sudden death from ischemic heart disease and toward even slight to moderate excesses of vitamin D as a risk factor in hyperlipidemia and myocardial infarction (Review: Linden, 1975/1977). Experimental data demonstrate that magnesium is protective against several models of myocardial disease, including those caused by hormonal and nutritional imbalances, stress, and hypoxia (Reviews: Seelig, 1972; Seelig and Heggtveit, 1974).
It is important to note that the proposal that magnesium deficiency is a contributory factor in cardiovascular disease does not negate the role of high fat intakes (which interfere with magnesium absorption). Also, the theories implicating the fatty streak as an early infantile atherosclerotic lesion do not preclude the theories that elastica degeneration is one of the earliest arterial lesions. Lipid droplets are seen in conjunction with damaged elastica (Duff and McMillan, 1951) and have been correlated with elastica degeneration (Pickering, 1963; Zugibe, 1963). Furthermore, elastica degeneration predisposes to lipid deposition (Kramsch et al., 1970, 1971). The papers that stress the changes in the musculoelastic layers of the arteries of infants as the earliest signs of arteriosclerosis support the premise that magnesium deficiency during the perinatal period, and factors that increase magnesium loss then and in early childhood, can contribute to the pediatric origins of cardiovascular disease, since comparable changes are seen in magnesium deficiency (Review: Seelig and Haddy, 1976/1980.
Because excess (saturated) fat has been considered a major contributory factor in atherosclerosis and ischemic heart disease, and experimental and epidemiologic studies implicate magnesium deficiency(infra vide), evidence of interrelationships between fat and magnesium is considered first.
5.1.1.1. Dietary Fat and Magnesium Balance
Evidence was obtained early that diets rich in fat interfere with magnesium absorption. In 1918, Sawyer et al., performed metabolic balance studies with 2 boys, 5 and 8 years of age, in which they explored the effects of fat intake on retention of calcium and magnesium. Even though their magnesium and calcium intakes were lower than in their normal diet, increasing the fat intake resulted in their excreting more of the divalent cations in both feces and urine. In a study of mineral balance of 4 young women on controlled magnesium-rich diets (800 mg/day), substitution of butter for vegetable fat resulted in retention of more magnesium (Bogert and Trail, 1922). On lower intakes of magnesium (320-350 mg/day), 6 young men given controlled diets containing 10-30% linoleic acid tended to be in negative magnesium balance while on the fatty-acid-supplemented diet (Irwin and Wiese, 1961). More extensive metabolic balance studies confirmed the interference by unsaturated fatty acids with magnesium retention in 19 young men on typical American intakes of magnesium, averaging 300-400 mg/day or 3.8-6.3 mg/kg/day (Hathaway, 1962). Magnesium, calcium, and phosphorus balances were calculated for ten 5-day periods, during which the diets were supplemented with linoleic acid at 9-10% and a subsequent increase in fatty acid to 20-30%. Most of the subjects were in probable magnesium equilibrium on the low-fat diets, considering balance to fall between 0 and + 18 [the sweat loss of magnesium, that averages 18 mg/day (Seelig, 1964) not being allowed for in the figures given (Hathaway, 1962)]. When the fat intake was increased, although the magnesium intakes remained approximately the same, most retained less magnesium (Fig. 5-1). Only 1 of the 19 men remained in strongly positive magnesium balance on the high fat intake. Of 11 who were in ± magnesium balance during the low fat intake, 6 showed essentially no change on high fat, and 5 went into negative magnesium balance. Two, who were in slightly positive magnesium balance on low fat, dropped to no retention on high fat. Of five who were in strong negative balance on the low-fat diet, four continued to lose substantial amounts of magnesium when the fat intake was increased, and one lost less. Thus, 11 of the 19 subjects lost more magnesium on the high- than on the low-fat diets.
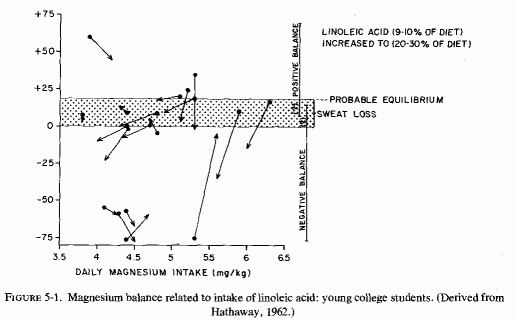{kind=link}
In a 5-day metabolic study of five normal medical students given a liquid diet, such as that given to preoperative peptic ulcer patients (that delivered 138 g of fat, predominantly from whole milk and cream, and that provided only about 200 mg of magnesium daily), there was an average daily loss of 16 mg of magnesium (Macbeth and Mabbott, 1964). This amount of dietary fat is equivalent to that of the typical American diet, comprising about 40% of the daily calories (de los Rios, 1961). This liquid diet differed from that given to the young men on the linoleic acid diet (Hathaway, 1962) in that the ratio of calcium to magnesium was 9:1, rather than 2:1.
Metabolic balance studies in rats on low- and high-fat diets have also shown that increasing the fat intake decreases the amount of magnesium absorbed from the gut (Olson and Parker, 1964; Tadayyon and Lutwak, 1969).
5.1.1.2. Steatorrhea and Magnesium Loss
Diseases that interfere with fat absorption, thereby resulting in high concentrations of fat in the intestinal lumen, interfere with magnesium absorption in several ways. Insoluble complexes of the fats and magnesium and calcium prevent their transit across the intestinal membranes, in conditions such as celiac disease or sprue, and in the steatorrhea that develops after gastrointestinal resections. [Bowel resections or bypasses further contribute to magnesium depletion by shortened transit time, decreased absorption area, and decreased enzymatic lipolysis (Opie et al., 1964).]
5.1.1.3. Dietary Fat and Blood Lipids (Man)
Despite the evidence that fat interferes with magnesium absorption, and that steatorrhea has caused hypomagnesemia and acute and subacute magnesium-depletion syndrome, the short-term studies of the effect of high fat intakes on serum magnesium have shown no effect For example, Macbeth and Mabbott (1964) who found that young men on a "Sippy-like" ulcer liquid diet for five days, who were in negative balance, maintained normal serum magnesium levels (1.9 ± 0.34 mEq/liter) Studies with older (42-62 years of age) schizophrenic patients on diets delivering 34, 65, and 134 g of fat (each patient given each of the diets for two-week periods in different sequences) showed no change in serum magnesium levels on the different diets though serum cholesterol levels fell on the low-fat diets (de Los Rios, 1961).
The epidemiologic studies of residents of hard- and soft-water cities, which consider the fat intakes, the magnesium and calcium content of the water supplies, and serum lipid and magnesium and calcium levels (Bierenbaum et al., 1973), provide interesting insight into the protective effect of hard water against sudden death from ischemic heart disease (supra vide). Comparison of these parameters in hard-water American and English cities (Omaha, Nebraska and London) with soft-water American and Scottish cities (Winston-Salem, North Carolina, and Glasgow) provides data that implicate the cations more than the fat ingested in the substantially lower cardiac death rates in Nebraska and in London than in the soft-water cities of southeastern United States and Glasgow (Review: Seelig and Heggtveit, 1974, and supra vide). For example, there was no significant difference in the percentages of those in Omaha and Winston-Salem (51.5% and 47.4%) who ingested diets high in fat, and in their serum cholesterol, triglycerides, and phospholipids. The tested residents of Glasgow, 72.8% of whom ate diets high in fat, had essentially the same serum lipid levels as did the tested London residents, only 28.2% of whom ate high fat diets. Nonetheless, the serum cholesterol and triglyceride levels of Glasgow residents were the lowest of all the four cities. Also, residents of both hard-water cities had higher serum cholesterol than did those of theft paired soft-water cities (p < 0.05). In evaluating the comparable serum levels of magnesium in both American cities, but the significantly higher serum magnesium in London than Glasgow, the noted common use of water softeners in Omaha (but not in London) should be considered. Residents of both American cities had significantly higher serum calcium levels (10.37 in Omaha, 9.59 in Winston-Salem) than did those in Britain (8.57 in London, 8.7 in Glasgow). The possibility that this is a reflection of more milk and vitamin D ingested by adults in the United States than in Britain should be considered, since the calcium content of London water was 2-3 times as high as that in the American cities.
With these data in mind, it is not surprising that there has been disagreement as to correlation of serum magnesium and cholesterol levels in patients with cardiovascular disease, or in populations at different risk.
5.1.1.4. Serum Magnesium and Cholesterol Levels in Cardiovascular Patients and High-Risk Populations
Bersohn and Oelofse (1957) and Bersohn (1958) correlated the lower serum cholesterol and slightly higher magnesium levels in Bantus than in white South Africans, with the lower incidence of arteriosclerosis and the higher dietary intake of magnesium of the Bantus. They analyzed the serum magnesium levels of Europeans with low to high serum cholesterol levels and found that, although there was overlap of serum magnesium values, the mean magnesium level of those with low serum cholesterol (mean = 170 mg/100 ml) was higher (1.7 mEq/liter) than was that of patients with hypercholesterolemia (cholesterol: 310-586 mg/100 ml; magnesium: 1.4 mEq/liter). In an Australian study comparing serum cholesterol and magnesium levels in several groups of aborigines and Europeans, Charnock et al. (1959) confirmed the lower serum cholesterol levels of the aborigines (who have a low incidence of cardiovascular disease) than of the Europeans and found significant differences (p = 0.001) between serum magnesium levels of the aborigines (1.7 mEq/ liter) and the Australians living in Adelaide (1.2 mEq/liter). Another group of Australians, living in a northern area (Alice Springs) one thousand miles away, had high mean serum magnesium levels (1.9 mEq/liter) and the highest mean serum cholesterol (314 mg/100 ml) of all the groups tested. Thus, the correlation between magnesium and cholesterol levels was not consistent. (The nature of the water of Alice Springs was not given.) It was interesting that there was no difference in serum magnesium (1.3 mEq/liter) and cholesterol levels (286; 281 mg/100 nil) in ischemic heart disease patients and age-matched European controls in Adelaide.
D. F. Brown et al. (1958), noting the report by Bersohn and Oelofse (1957) (supra vide) and that by Malkiel-Shapiro et al. (1956) that parenteral administration produced clinical improvement and lowered β-lipoprotein levels in patients with myocardial infarction (MI), studied serum magnesium-lipid relations in MI patients and in middle-aged controls. They found no correlation between serum magnesium and lipid levels, and no significant difference between the patients and the controls. Similar negative findings have been reported by others in studies of patients with cardiovascular disease and hyperlipidemia (Hyatt et al., 1966; Murnaghan et al., 1969; Rotman et al., 1971/1973).
On the other hand, Jankelson et al. (1959), who compared serum magnesium and lipid fractions of atherosclerotic patients and controls, found that although total cholesterol levels were the same in both groups, there were differences in magnesium and lipoprotein levels. The avenge serum magnesium level was 1.4 mEq/liter in 23 atherosclerotic patients and 1.6 in 12 healthy controls (in third and fourth decades of life). The β-lipoproteins were 11.5 in patients and 8.5 in controls; the α-lipoproteins were 5.3 in patients and 2.2 in controls. Six of the atherosclerotic patients were alcoholics; all had normal cholesterol levels, but 4 had high β-lipoprotein and all had higher than control α-lipoprotein values; 4 had very high levels. There was not good correlation, however, of low serum magnesium levels with high lipoproteins. Three with arteriosclerotic heart disease, and/or cerebral thrombosis, respectively, had hypomagnesemia (0.7, 0.7, and 1.3 mEq/liter) and hyper-β-lipoproteinemia (8, 13.4, and 9). But 2 with comparable disease and high β-lipoproteins (12.5 and 14.3) had normal serum magnesium (1.7 and 2.0 mEq/liter). One with cerebral thrombosis had normal magnesium and lipid levels. High serum cholesterol levels (288 mg/100 ml) and low serum magnesium levels (1.5 mEq/liter) were seen in 25 patients with acute MI a week after the infarction, as compared with the avenge levels in 50 controls (cholesterol: 210; magnesium: 2.1 mEq/liter) and in 15 old MI cases (cholesterol: 238; magnesium: 1.9) (Nath et al., 1971/1973). The magnesium levels rose during the next two weeks to normal (1.9 mEq/liter). Patients with angina pectoris, in this series, had high cholesterol levels (278) but normal serum magnesium values (2.0 mEq/liter). Rangam and Gupta (1961) found that among 44 patients with hypercholesterolemia, 80% had hypomagnesemia; among 52 with high lipid phosphorus levels, 75% had low serum magnesium levels. Those with normal cholesterol levels, however, also had a high incidence (54%) of hypomagnesemia in this series.
A brief abstract reports highly significant (p = .001) correlations between magnesium and high cholesterol and low-density lipoproteins in a survey of 32 random subjects 40-60 years of age (Mondschein, 1974). Over half of the magnesium values were below the normal range; none was above.
Many of the attempts to determine whether high serum cholesterol levels correlated with low magnesium levels in patients with cardiovascular disease derived from the clinical reports that parenteral magnesium administration was of value in treating patients with myocardial infarcts (MI), coronary insufficiency, and/or peripheral arteriosclerosis. Malkiel-Shapiro et al. (1956) first reported lowering β-lipoproteins in patients with coronary insufficiency, with the use of intramuscular (i.m.) MgSO4 begun at the time of an acute attack of coronary thrombosis or during acute coronary insufficiency. The regimen employed by Malkiel-Shapiro (1958) for 25 years involved deep i.m. injections of 0.5-2.0 ml of 50% MgSO4 on alternate days, and at longer intervals (to twice weekly) as the condition improved. Patients who had recently recovered from an MI or who suffered angina of effort were usually given 12 i.m. injections of MgSO4 at 5-day intervals, which was repeated after 4-6 months if they had benefited. These physicians stated that patients with more advanced disease seemed to have the most striking improvement. In the earlier studies (Malkiel-Shapiro et al., 1956), patients on magnesium therapy were given no anticoagulants. Subsequently, concurrent use of small doses of heparin (5000 units daily) and i.m. MgSO4 (0.5 ml 50% solution) once weekly were found useful in the maintenance of patients who have recovered from an acute MI (Malkiel- Shapiro, 1958; Malkiel-Shapiro et al., 1960). After the 1956 report of this group, clinical trials were undertaken in South Africa (Teeger, 1958; Agranat 1958; Marais, 1958) and Australia (R. Parsons, 1958; R. Parsons et al., 1959, 1960, 1961), and there was clinical verification of much that had been claimed. Agranat (1958) reported a 44% improvement rate with the use of MgSO4 injections to patients with chronic IHD. R. Parsons (1958) reported briefly that 3 injections of 1 ml 50% MgSO4 weekly for a month to IHD patients resulted in reversal of low lecithin/cholesterol ratio, lowering of the β-lipoprotein levels with elimination of the pre-β- band, and reduction of plasmin inhibition. In a detailed report, R. Parsons et al. (1959) described treatment of patients with angina but no ECG evidence of MI, and of patients with ECG evidence of MI with or without angina. They found that 2-ml doses of 50% MgSO4 given i.m. every 5 days (until 12 doses were given), were more effective than were the 1-ml doses. Patients with acute MI were also given heparin for the first 3 days of treatment. Comparison of the results of this regimen with that obtained the previous year when only anticoagulants were used were striking. Of over 100 patients given the magnesium therapy, one-third of whom had had acute MI, there was only one death. Among almost 200 patients treated with anticoagulants alone, 60 died. The biochemical changes (Table 5-1) show the improvement in lecithin/cholesterol ratio, the decrease-particularly in β-lipoproteins-and in plasmin inhibition produced by the magnesium therapy. In 1960, Parsons et al. published confirmation of the observation (Malkiel-Shapiro, 1958) that combination of low dosage heparin with i.m. magnesium therapy was even more effective in speedily reducing β-lipoproteins and total lipids to normal levels. They recommended that patients with acute MI should be given heparin (15,000 units every 6 hours for 3 days), with an initial dose of 2 ml 50% MgSO4 i.m. Then low-dosage (5000 units) heparin and 2 ml 50% MgSO4 were given three times weekly for 6 weeks, and once weekly subsequently.
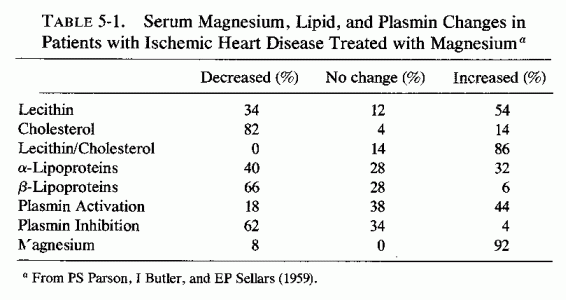{kind=link}
Application of this regimen to patients with angina, MI, or peripheral arterial disease (incipient gangrene, ischemic leg ulceration, Raynaud's disease, and intermittent claudication) has been reported to produce clinical improvement and to lower serum cholesterol levels (S. Browne, 1961, 1963, 1964a,b). Savenkov et al. (1971) have also reported that treatment with a preparation containing magnesium adipate and magnesium nicotinate (in tablet or ampoule form for i.v. or i.m. administration) has been useful in the treatment of 54 patients with coronary, cerebral, and (in 21 cases) peripheral atherosclerosis. Treatment was given for 20 days parenterally (18 patients), orally (20 patients), or parenterally for half the course, followed by oral administration (16 patients). The clinical response was considered good in 22 instances, satisfactory in 16, and effective in 10. The total serum cholesterol was obtained in 41 patients (average = 284 ± 16.9 mg/100 ml). The level decreased in 29 patients, did not change in 9, and rose by 24 mg/100 ml in 3. In the entire group, there was an average decrease of 17.5%.
A magnesium-aluminum-siliconate preparation was given in fairly high dosage (2-3 g/day) to hyperlipemic patients without (Table 5-2A) and with (Table 5-2B) moderate to severe manifestations of cerebral, coronary, or peripheral arterial disease (Lieber, 1961). The most reduction was in the esterified cholesterol fraction, an interesting finding in view of the high levels of cholesterol esters in magnesium-deficient dogs (Kruse et al., 1933) and rats (Savoie and Delorme, 1976/1980). The ratio of β/α-lipoproteins fell increasingly with the duration of administration of the magnesium preparation, and to a greater degree in group A than group B. The low-dosage magnesium nicotinate preparation was given to patients with lesser degrees of hyperlipidemia, and produced less striking changes, but in the same direction (Table 5-1C, D) (Lieber, 1961). The tendency toward magnesium-induced decreased β-lipoprotein levels is reminiscent of comparable findings in magnesium-supplemented rats on atherogenic diets (infra vide).
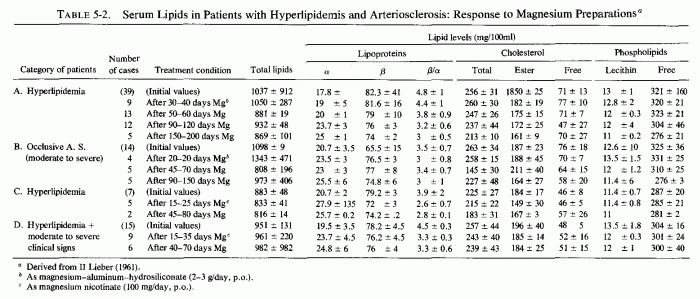{kind=link}
A brief abstract of a long-term (19-month) double-blind study of 35 patients given either oral MgCl2 and KCl (1 mEq/kg/day) or placebo, reported that serum β-/α-lipoproteins were 10% lower in the treated group than in the placebo group (Haywood and Selvester, 1962). The dose-limiting effect of side effects was considered a likely explanation of the failure to reduce the lipids further. Smaller doses of magnesium given in complexed or chelated form were reported to lower the elevated β-fraction somewhat, but not to lower the total cholesterol levels; when the magnesium was stopped the β-fraction rose to pretreatment levels (A. Steiner, 1962, 1963). In the latter study, β-vitamins were also given.
Rademeyer and Booyens (1965), having shown that the maize meal dietary constituent of the Bantus had a hypocholesterolemic effect in rats, which they attributed to its high magnesium content and to its interference with fat absorption, demonstrated that supplementation with maize meal of diets of hyperlipemic whites raised their serum magnesium and lowered their serum cholesterol levels (Booyens et al., 1966).
The conflicting clinical reports as to the relationships of serum magnesium and lipid levels, and their meaning in cardiovascular disease, call for evaluation of magnesium-lipid interrelationships in experimental atherogenesis. In particular, because of the poor correlation between serum lipid levels and protection against heart disease in hard-water areas (Bierenbaum et al., 1973), and because magnesium administration has been claimed to be beneficial in overt clinical cardiovascular disease, animal data deserve careful scrutiny.
Among the numerous studies of atherosclerosis induced by fat, cholesterol, and cholic-acid-loading of animal diets, few have included determination of magnesium levels. The early magnesium-deficiency studies by Kruse et al. (1933) showed that young dogs on an atherogenic diet low in magnesium (.08% of diet) and containing butter fat (8% of diet), exhibited no change in total blood lipids, little or no change in free cholesterol, a drop in fatty acids, but a substantial rise in esterified cholesterol. As the magnesium level dropped, the percentage of esterified cholesterol rose. The young dogs on a magnesium-free, but otherwise well-balanced, diet, which delivered no lipid other than corn oil, were found by Vitale et al. (1961) to develop neither elevation of blood cholesterol nor atheromatous plaques. Bunce et al. (1962a,b) demonstrated that increasing the magnesium intake from 80 to 180 ppm, in dogs fed 20%-animal-fat diets, prevented the aortic lesions seen in dogs on the lower magnesium intake, but allowed for a slight further rise in serum cholesterol.
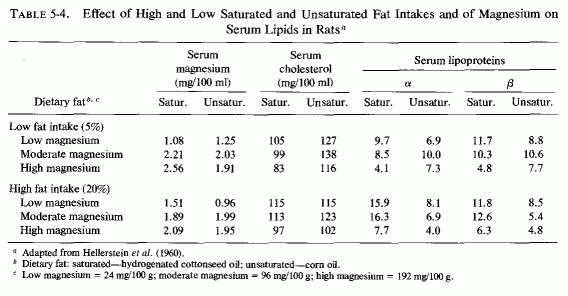
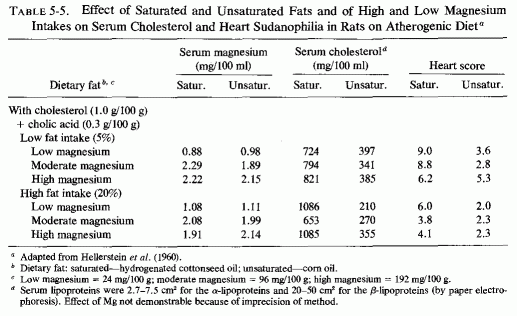
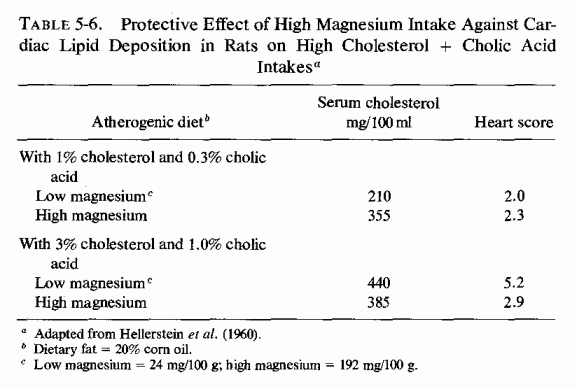
The atherogenic diet fed to rats (Vitale et al., 1957a,c,d,e; Hellerstein et al., 1957, 1960) produced marked hypercholesterolemia (639-808 mg/100 ml) that was not lowered by increasing the magnesium intake, even though early arteriosclerotic lesions were diminished (Vitale et al., 1957d,e; Nakamura et al., 1960). In rats on the atherogenic diet, also high or low in protein (Vitale et al., 1957c) or calcium (Vitale et al., 1959), increasing the magnesium content caused a further rise in serum cholesterol. A high intake of both magnesium and calcium, reduced the sudanophilia of the hearts to 4.0 from the high value of 8.3, but exerted little influence on serum lipids (Table 5-3). Increasing the magnesium intake of rats on low calcium intake substantially lowered the β-lipoproteins. A high magnesium intake slightly lowered the serum cholesterol and more profoundly lowered the lipoproteins of rats on high and low fat intakes, whether the fats were saturated or unsaturated (Hellerstein et al., 1960: Table 5-4). No cardiac sudanophilia developed, unless cholesterol and cholic acid were added to the diet. The markedly elevated plasma cholesterol, seen in rats also given cholesterol and cholic acid, was actually increased on the higher Mg intakes, although the cardiac lipid deposition in the rats on saturated fats and high Mg was reduced. Altering the Mg intake did not notably affect the lesser heart scores of rats on high intakes of unsaturated fat (Table 5-5). The elevation of heart scores of rats on low unsaturated fat diets when their magnesium intake was increased requires elucidation. Vitale et al. (1959) and Hellerstein et al. (1957, 1960) suggested that magnesium might protect against lipid deposition in the cardiovascular system by means of its effect on lipoprotein metabolism. They demonstrated that further increasing the intakes of cholesterol to 3% and cholic acid to 1%, of rats on 20% unsaturated fat, increased serum cholesterol levels only slightly (to 440 mg/100 ml), but increased the heart scores of rats on low magnesium intake to 5.2. High magnesium intake protected against this increased heart score (Table 5-6). Increasing the magnesium intake of rats on atherogenic diets, given alcohol or water to drink, also resulted in higher serum cholesterol levels, but less cardiovascular sudanophilia (Vitale et al., 1957a). Nakamura et al., (1960, 1966) showed that the long-term feeding of 192 mg/100 g of magnesium to rats on this atherogenic diet produced an early increase in serum lipids that fell only gradually within the year-long observation, but a significant decrease in arterial lipid deposition was evident within two months on the magnesium-supplemented diet.
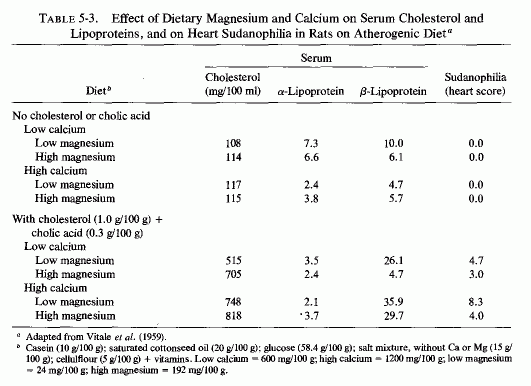{kind=link}
{kind=link}
{kind=link}
{kind=link}
In contrast to the results in the foregoing studies with hypercholesterolemic semisynthetic diets, the high blood cholesterol produced in rats fed whole milk (containing 4 g butter fat/100 ml milk) alone or with added cholesterol, was corrected by adding MgSO4 to the diet (Mullick and Kakkar, 1963). It seemed possible that formation of insoluble compounds of the milk fat and magnesium might have prevented absorption of the excess fat. However, in another report, magnesium salt given intramuscularly also lowered the serum cholesterol (Kakkar and Mullick, 1963).
Rademeyer and Booyens (1965) explored the effect of butter fat versus sunflower-seed oil on the serum magnesium and cholesterol levels of rats fed a semisynthetic low magnesium diet similar to that used by Vitale's group (supra vide). They found that the addition of 25% butter fat to the diet lowered the serum magnesium from 3.3 to 2 mEq/liter and raised the serum cholesterol from 65.8 to 81.6 mg/100 ml over a 4-week period (p = 0.001). The serum magnesium did not fall on addition of 25% sunflower-seed oil, nor did the serum cholesterol rise. An equal amount of meat-fat drippings caused a lesser fall in serum magnesium than did the butter fat, and lesser but significant rise in serum cholesterol. Substituting sunflower-seed oil for butter in the group that had been fed the butter-supplemented diet for 4 weeks affected neither the depressed serum magnesium nor the elevated serum cholesterol, but substitution of maize meal for glucose caused a rise in magnesium and a fall in cholesterol within a week. Maize meal (a major dietary constituent of Bantus) was used in this study in an effort to determine why Bantus have a lower serum cholesterol and higher serum magnesium level, as well as a lower incidence of arteriosclerosis than do South African whites (Bersohn and Oelofse, 1957).
Hungerford and Bernick (1976/1980) have recently reaffirmed the lack of alteration of plasma magnesium in rats on synthetic atherogenic diets, and elucidated the histologic arterial changes produced by an atherogenic or magnesium-deficient, or combined high-fat low-magnesium diet. They showed the further increase in serum cholesterol produced when rats on atherogenic diets were also magnesium deficient.
Rabbits on a hypercholesterolemic diet for 24 weeks showed a sharp drop in serum magnesium (and calcium) at 6 weeks. The hypomagnesemia persisted for 6 more weeks and then tended to rise (Rangam and Gupta, 1961). Intravenous MgSO4 injection (2 ml 5% solution) to such rabbits was found to lower serum cholesterol for 48 hours (Rangam and Gupta, 1962). Magnesium deficiency intensified the deposition of fat in the aortas of rabbits on atherogenic diets, lowered the level of serum triglycerides significantly (p = 0.05), but exerted little effect on total serum cholesterol (Nakamura et al., 1965). Magnesium supplementation had little effect on serum aorta lipid levels in rabbits in one study (C. Adams et al., 1964). Neal and Neal (1962) found higher serum phospholipid and triglyceride levels in rabbits on atherogenic diet when their drinking water contained magnesium than when they were given distilled water to drink, but they had less atherosclerosis when they were magnesium supplemented. Another group confirmed these observations. They found that administration of magnesium (as Mg Na2 EDTA) had little effect on the hyperlipidosis of rabbits on atherogenic diet but reduced formation of atheromatous plaques (McCann et al., 1962; Wartman et al., 1967). The magnesium-deficient cebus monkeys on atherogenic diets, reported by Vitale et al., (1963), showed both elevated serum cholesterol values and marked intimal lipid deposition in the aorta, not seen in controls. A study of the response to [3H]cholesterol given intravenously to magnesium-deficient and control rats, showed that the tagged cholesterol was taken up and subsequently released more rapidly by the liver of magnesium deficient than control rats. As a result, there was an initially greater drop in serum [3H]cholesterol and a greater subsequent rise in the magnesium-deficient rats; they also exhibited extracellular [3H]cholesterol between the elastic lamellae and the smooth muscles in the aorta (Schmalbeck et al., 1972).
The Mg- and KCl-free diet, containing animal fat, vitamin D, and sodium phosphates, which was contrived by Sos et al. (1964a,b,c) to be cardiopathogenic in several species, produced elevated serum cholesterol levels (Review: Seelig and Haddy, 1976/1980). A similar diet, designed to be thrombogenic, but that also produced cardiac necrosis in rats (Savoie, 1972a,b, 1975; Savoie et al., 1973), produced a substantial rise in blood cholesterol levels, particularly in the esterified form (Savoie and Delorme, 1976/1980), a finding that recalls the early observation by Kruse et al. (1933) in dogs. Blood phospholipids were also increased, and blood magnesium levels were lowered. Magnesium supplementation of the atherogenic or of the thrombogenic diet exerted little effect on most of the blood lipid fractions, raising some further and lowering some slightly but none to normal levels (Savoie and Delorme, 1976/1980). It is noteworthy that hypocholesterolemic agents (clofibrate, nicotinic acid, and conjugated estrogens) exerted no protective effect against the nonocclusive suppurative cardiac necrosis produced when Na2 HPO4 was added to the hyperlipemic thrombogenic diet. Only MgCl2 was completely protective (Savoie, 1972b). Further work showed that the sodium phosphate addition accentuates the hypokalemia of the thrombogenic diet, but produces hypomagnesemia, and lowers the cardiac magnesium levels. More recently, Savoie and Delorme (1976/1980) found that the thrombogenic diet increased lipoprotein lipase activity, an effect not influenced by magnesium. On the other hand, the added phosphate lowered the cardiac lipase activity, and magnesium raised it, with resultant elevation of cardiac free fatty acid levels. Magnesium lowered the free cholesterol levels in the hearts of the rats on the cardiopathic diet.
Catecholamines have long been known to increase the blood levels of free fatty acids, whether by injection in animals (Dole, 1956; Bogdonoff et al., 1961) or as a result of such stress as myocardial infarction (Kurien and Oliver, 1966; Kurien et al., 1971; Oliver et al., 1968; Editorials, Lancet, 1969a,b). The complex interrelationships of magnesium and catecholamines and corticosteroids have been surveyed by Wallach (1976/1980) and those of corticosteroids (which are also released by stressful situations) and magnesium by Massry and Coburn (1973). In the case of the catecholamines, depending on the time of testing and the test situation, they have both increased and decreased blood magnesium levels and decreased tissue (i.e., heart) magnesium levels.
Considered here are the blood lipid-magnesium interrelationships, as influenced by cold-stress, the acute alcohol-withdrawal syndrome, and the administration of catecholamines, which might provide some insight into the somewhat contradictory findings. Rayssiguier and Larvor (1976/1980) have recently reported that either fasting or exposure of shorn young sheep to cold temperatures causes comparable lipolysis to that produced by infusions of either epinephrine or theophylline. Hypomagnesemia accompanied the increase in free fatty acid levels in the blood, caused by each of the stimuli. Sodium nicotinate, which is antilipolytic, inhibited both the increase in free fatty acids in the blood and the decrease in blood magnesium. High blood levels of long-chain free fatty acids are also seen during the acute phase of alcohol withdrawal (Mays et al., 1970) and the severity of the symptoms tends to be greater in those with higher levels of the fatty acids (Blink et al., 1973, 1976/1980). Since such fatty acids can chelate magnesium, Flink et al., (1976/1980) propose that the signs of alcohol withdrawal may depend upon inactivation of magnesium by the fatty acids. They verified the elevation of free fatty acids in the blood of dogs induced to imbibe alcohol and suggested that reducing lipolysis during alcohol withdrawal might be useful in controlling the symptoms, which are often controllable by magnesium repletion (Rink et al., 1954, 1957; Rink, 1956, 1969, 1976/ 1980)
The mobilization by catecholamines of free fatty acids and their inactivation of magnesium recall Browne's early (1964a,b) deduction that the clinical benefit reported from magnesium therapy of hyperlipemic patients with occlusive arterial disease might derive from magnesium-catecholamine interrelationships. He pointed out that magnesium inhibits catecholamine release from the adrenal medulla (Douglas and Rubin, 1961, 1963, 1964), and that smoking or nicotine infusions (to dogs) causes elevation of both serum free fatty acid levels and urinary excretion of catecholamines (Kershbaum and Bellet, 1964). The arrhythmia following clinical myocardial infarction might be related to the catecholamine-induced increase in circulating fatty acids that might be mediated by inactivation of serum magnesium. Perhaps more likely is the possibility that increased myocardial lipids, such as have been attributed to catecholamine lipid mobilization in rats injected with sympathomimetic agents (Ferrans et al., 1964, 1969) and in electrolyte-steroid cardiopathy (Prioreschi, 1966), might be the result of inactivation by the intramyocardial fats of cellular magnesium. It is provocative, in this regard, that a direct correlation was made by Balazs et al., (1962) with the cardiotoxicity of isoproterenal in rats and the amount of excess body fat. The availability of more fat for lipolysis under stressful situations might explain the greater susceptibility of obese individuals to fatal ischemic heart disease.
The treatment of men with coronary insufficiency by estrogens is no longer advocated. Estrogens used to be administered in an effort to lower the β/α-lipoprotein ratio of these men to that of young women (Ban, 1955; Oliver, 1960) because of the sex difference in the incidence of ischemic heart disease. Despite success in lowering the β- and raising the α-lipoprotein levels by giving estrogens (Barr et al., 1952; Townsend et al., 1952; Gertler et al., 1953, Steiner et al., 1955; R. W. Robinson et al., 1956; Voyles and Evans, 1961), there has not been satisfaction that a sufficiently suppressive effect is exerted on recurrence of cardiovascular accidents to justify the unpleasant side effects (Steiner et al., 1955; Oliver, 1962; Robinson et al., 1963).
The effect of estrogens on blood coagulation may provide a possible explanation of their failure to achieve benefit in patients who had suffered a myocardial infarction, a condition associated with increased coagulability of the blood shortly after the event (McDonald and Edgill, 1957, 1959; Katzet al., 1963). Estrogens have long been known to increase the coagulability of blood, an attribute that has been used to stop bleeding [e.g., after tonsillectomies (S. Fox, 1960) and to control epistaxis (E. Blackburn, 1963)]. This activity, however, seems relevant to the correlation of thromboses and infarctions with the use of estrogen-containing oral contraceptives (Inman et al., 1970; Coronary Drug Project Report, 1973a; Editorial, Lancet, 1977; Goldsmith and Johnston, 1979).
If one accepts the premise that formation of mural thrombi is a pathogenic mechanism in atherogenesis (Duguid, 1946; T. Crawford, 1959; Astrup, 1959; McDonald, 1959; Pilgeram, 1961; Pickering, 1963. A. Katz et al., 1963), the enhancement of intravascular coagulation by estrogens should result in a higher, rather than the lower incidence of cardiovascular disease in premenopausal women than in men.
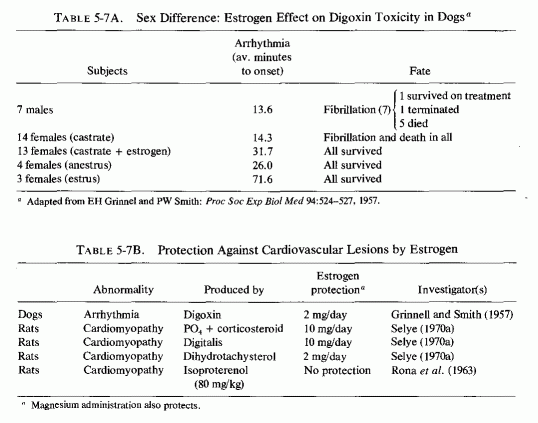
It was speculated (Seelig, 1964) that the common denominator between the low incidence of ischemic heart disease in men from the Orient (where the intake of saturated fat is low) and in young women might be magnesium. The substantially higher dietary intakes of magnesium in the Orient, and the better retention of magnesium by young women than young men on the customary marginally adequate magnesium intake of the Western world, suggested that the adequacy of magnesium might be the protective factor against IHD. If the effective retention of magnesium by women reflects its affinity, not only to target tissues such as those involved in the reproductive process and to bone (Walaas, 1950; Csapo, 1956; Best and Pickles, 1965; N. Goldsmith, 1971) but to the cardiovascular system, that might elucidate the greater resistance of premenopausal women than of older women and of both young and older men to cardiovascular disease. Thus far, experimental data verifying the greater resistance of females than males to cardiopathic agents have not elucidated the possible role of cardiovascular magnesium levels. One can draw some inferential conclusions, but definitive work remains to be done. For example, female dogs and rabbits are more resistant than are males to digitalis-induced arrhythmias (Grinnell and Smith, 1957; Rodensky and Wasserman, 1964). Castrated females are as susceptible as are males to digitalis toxicity; estrogen replacement (2 mg/day) markedly improves their resistance to arrhythmias, but not to the extent seen in estrus (Table 5-7A, Grinnel and Smith, 1957). High-dosage estrogen has been almost fully protective against the myocardial necrotic lesions produced by phosphate and corticoids and by digitalis overdosage and has protected against myocardial necrosis caused by dihydrotachysterol (Table 5-7B, Selye, 1970a). It is thus of interest that digitalis toxicity is increased by magnesium deficiency (Vitale et al., 1963), that digitalis increases the renal excretion of magnesium (Kupfer and Kosofsky, 1965), and that magnesium is useful in digitalis toxicity (Zwillinger, 1935; Szekely and Wynne, 1951; J. Stanbury and Farah, 1960; Cook et al., 1967; Wacker and Parisi, 1968; Seller et al., l970a,b). Although Ronaet al. (1963) could not protect against the myocardial necrosis (produced by massive doses of isoproterenol) with estrogens, female rats are more resistant than are males to this form of cardiac damage, an effect that Rona et al. (1963) attributed to the slower rate of growth of the females. Possibly, the protective effect of estrogens in these experimental models might be mediated by increased uptake of magnesium by the myocardium, as well as by other tissues, in response to estrogen. Additional evidence that estrogen, or other female sex hormones, might be protective against several forms of cardiovascular lesions derives from study of the influence of pregnancy on experimental models. Advanced pregnancy in rats has protected against: (1) dihydrotachysterol-induced arteriosclerosis (Selye, 1957); (2) the cardiovascular necrosis and calcification and calcification of vitamin D excess (Potvliege, 1962); phosphate + corticoid-induced cardiomyopathy (Selye, 1958a); and hyperparathyroid myocardial necrosis (Lehr and Krukowski, 1961a,b; Krukowski, 1961, 1963; Lehr, 1965b). Pregnant dogs are more resistant than are nonpregnant females to necrotizing arteritis produced by a high-fat diet and renal insufficiency (Holman and Jones, 1953).
{kind=link}
It is possible that the paradoxical effects of estrogen on diseases of the cardiovascular system relate partially to its effects on magnesium distribution. It has been shown that serum magnesium falls with the cyclic increase in estrogen secretion (Dahl, 1950; Nida and Broja, 1957; Goldsmith, 1963; Goldsmith et al., 1970; Goldsmith, 1971). The use of estrogen-containing oral contraceptives has been shown to reduce the serum levels of magnesium (in users versus nonusers) by 16% (Goldsmithet al., 1966), 28% (DeJorge et al., 1967), and by 27% and 33% (Goldsmith, 1971). Evaluation of different contraceptives suggests that it is the estrogen moiety that is responsible for the decrease in serum magnesium (Goldsmith and Goldsmith, 1966; Goldsmith et al., 1970, Goldsmith and Johnston, 1976/1980) although there are conflicting findings. Since rats given estrogen showed decreased serum magnesium levels, without increased urinary magnesium output, and since the bone-magnesium increased, Goldsmith and Baumberger (1967) proposed that a shift of magnesium to the tissues was responsible for the estrogen-induced fall in serum magnesium. Indirect support for the importance of the estrogen component of contraceptives in lowering serum magnesium comes from the report that progestogens increase rather than decrease serum magnesium (Dale and Simpson, 1972). Yet, norethisterone and mestranol, alone or combined, have been shown to increase magnesium levels in bone, muscle, and intestinal wall tissues (Gozan and Charnot, 1973; Charnot et al., 1974). Despite the increase in tissue levels of rats on mestranol, their serum magnesium levels did not fall; norethisterone, however, produced a 30% drop in serum magnesium (Gozan and Charnot, 1973). The picture is further confused by the studies showing no effect of several oral contraceptives on serum magnesium (N. Hahn et al., 1972) or on magnesium levels of plasma, erythrocytes, and platelets (Thin, 1971). Data on decreased serum magnesium levels during pregnancy are discussed elsewhere in this volume, as possibly reflecting a true magnesium deficit rather than a hemodilution or estrogen-induced effect. Wallach (1976/1980) has considered the findings relating to the effect of estrogen on magnesium and has commented that circumstantial evidence from studies of interrelations of estrogen, calcium, and magnesium on thymic cell proliferation (Morgan and Perris, 1974) suggests that estrogen may favor cellular transport of magnesium.
Although there is no uniform agreement that estrogens lower serum magnesium levels, most of the evidence points in that direction. Thus, the still controversial evidence that low magnesium levels can contribute to coagulopathy deserves consideration as a possible factor in estrogen-induced thrombotic disorders. Durlach (1967a,b,c) first described severe thromboembolic disease in a young woman with latent tetany of magnesium deficiency. Her disorder was associated with increased ADP-induced platelet aggregation. Additional instances have since been reported in women with latent tetany of magnesium deficiency (DuPont et al., 1969; Durlach, 1970; Boudet et al., 1972; Erödi, 1973; Debrand, 1974; Maurat et al., 1974; Seelig et al., 1976/1980). Durlach (1970) has also shown that estrogen therapy gives rise to both functional platelet alterations and to signs of magnesium deficiency, which regress on administration of oral magnesium in moderate dosage. Vajna (1971/1973) has claimed that administration of magnesium to women on oral contraceptives significantly reduces the risk of coagulopathy.
Elin (1976/1980) and Durlach (1976/1980) have reviewed the in vitro evidence that magnesium plays a role (predominantly inhibitory) in the coagulation process. However, as Durlach (1976/1980) stresses, most of the in vitro studies showing that magnesium can inhibit coagulation factors-prothrombin, thrombin, and Factors V, VII, and IX-and can increase fibrinolysis, have been based on studies with high magnesium concentrations. They are thus not directly relevant to consideration of the effects of low or marginally low serum magnesium levels on the tendency toward intravascular coagulation. A few experimental magnesium-deficiency studies may shed light on the clinical coagulopathy of magnesium deficiency or on that accompanying use of agents (such as estrogens) that lower serum magnesium levels. Stevenson's and Yoder's (1970) magnesium-deficient animals had significantly shorter thrombin clotting time and greater ADP breakdown than did the normal group (p = 0.001). The partial thromboplastin time was also significantly reduced in magnesium-deficient calves (p = 0.05). Stachura (1971) observed hypercoagulation with shortened thromboplastin time in magnesium-deficient rats. Magnesium-deficient calves had insignificantly increased ADP-platelet aggregation (p = 0.05) but magnesium-deficient rats had more ADP-platelet aggregation (p = 0.05) than did normal rats (Stevenson and Yoder, 1970). Since magnesium-deficient rats commonly develop hypercalcemia (Larvor and Durlach, 1971a; Seelig and Haddy, 1976/1980),the species difference in ADP platelet response to magnesium deficiency might reflect the presumed difference in the Mg/Ca ratio in the rats versus the calves (data on calcium levels were not provided).
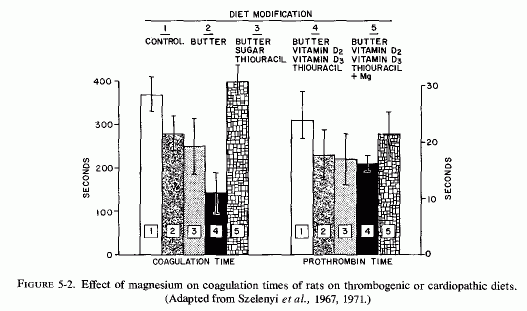
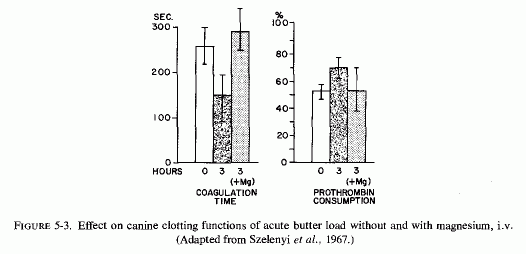
Hypercoagulability, produced by feeding rats a thrombogenic diet containing cholesterol, vitamins D2 and D3 and thiouracil, as well as large amounts of butter, was counteracted by oral magnesium chloride (Szelenyi et al., 1967) (Fig. 5.2). Dogs acutely loaded with butter showed markedly increased blood coagulability three hours later. Magnesium, given intravenously at the time of intragastric butter administration, prevented the decreased coagulation time and the increased prothrombin consumption (Szelenyi et al., 1967) (Fig. 5-3).
{kind=link}
{kind=link}
Clinical studies of the effects of magnesium administration to patients with cardiovascular disease and hyperlipidemia have been considered. Relevant to this section are the reports that parenteral magnesium therapy reduced plasmin inhibition (R. Parsons, 1958). Other studies suggest that magnesium therapy accelerates fibrinolysis (Hackethal, 1949; Zahnert and Oloffs, 1960). Further clinical investigation of effects of magnesium on blood coagulation and clot lysis is required.
Elin (1976/ 1980) has reviewed in vitro evidence that magnesium affects platelet aggregation and release. Its inhibitory effects on platelet aggregation have been with high concentrations (Born and Cross, 1964); the calcium/magnesium ratio is important at low concentrations (Herrman et al., 1970). Platelet release is calcium dependent (Sneddon and Williams, 1973); increasing concentrations of magnesium are inhibitory (Sneddon, 1972).
Whether these findings are relevant to the increased blood coagulability and platelet adhesiveness of patients with myocardial infarctions remains to be resolved. Hughes and Tonks (1965) reported significantly decreased serum magnesium levels and increased platelet aggregability in infarct patients, as compared with matched controls, a finding reported also by Prakash et al. (1971/1973). Chadda et al. (1976/1980) have also reported decreased serum magnesium in such patients. However, Murnaghan et al. (1969) reported elevated serum magnesium levels and Khan et al. (1974) normal levels, the latter in association with highly significantly increased platelet adhesiveness. In view of the known stress and anoxia-induced magnesium egress from the tissues, with initially increased serum magnesium, followed later by decreased serum magnesium levels, longitudinal studies of infarction patients must be done with meticulous attention paid to the time lapse after the ischemic event; and to the degree of decompensation-hypoxia.
Part II: Chapter 6
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
The major emphasis of the preceding sections on magnesium-lipid interrelationships and on estrogen-lipid/coagulation-lipid interrelationships is predominantly on blood constituents, as they influence the development of atherosclerosis.
The likelihood that metabolic and structural alterations in arterial walls may predispose to their increased accumulation of lipids has also been investigated. The need to consider, not only alterations in the constituents of blood but also in the status of the containing vessels, was commented upon by Duff and McMillan (1951) in their review of changing concepts of the pathogenesis of arteriosclerosis. They observed that the view that chemical and physicochemical aberrations of the serum lipids and lipoproteins are fundamental to the pathogenesis of arteriosclerosis had become so popular that "… the casual reader of recent literature might wonder whether some authors conceive of an atherosclerosis so independent of the substrate of the vessel wall, that it may occur in the absence of the blood vessels themselves."
Specific alterations in the mucopolysaccharides have been observed in the ground substance of arteriosclerotic arteries obtained from human material. Increased metachromasia, due to elevation in acid mucopolysaccharides, occurs in arteries from human material prior to lipid infiltration in aging and in arteriosclerosis (Faber, 1949; Moon and Rinehart, 1952; Moon, 1959; Gresham et al., 1962). It has been suggested that it develops in areas characterized by prior degeneration of the elastica and predisposes to infiltration by lipids (Moon and Rinehart, 1952; Taylor, 1953; Moon, 1957, 1959). On the other hand, it has been postulated that lipids in arterial lesions derive from the degenerated elastic fibers and that the elevation in mucopolysaccharide reflects a healing process (Zugibe and Brown, 1960; Zugibe, 1963).
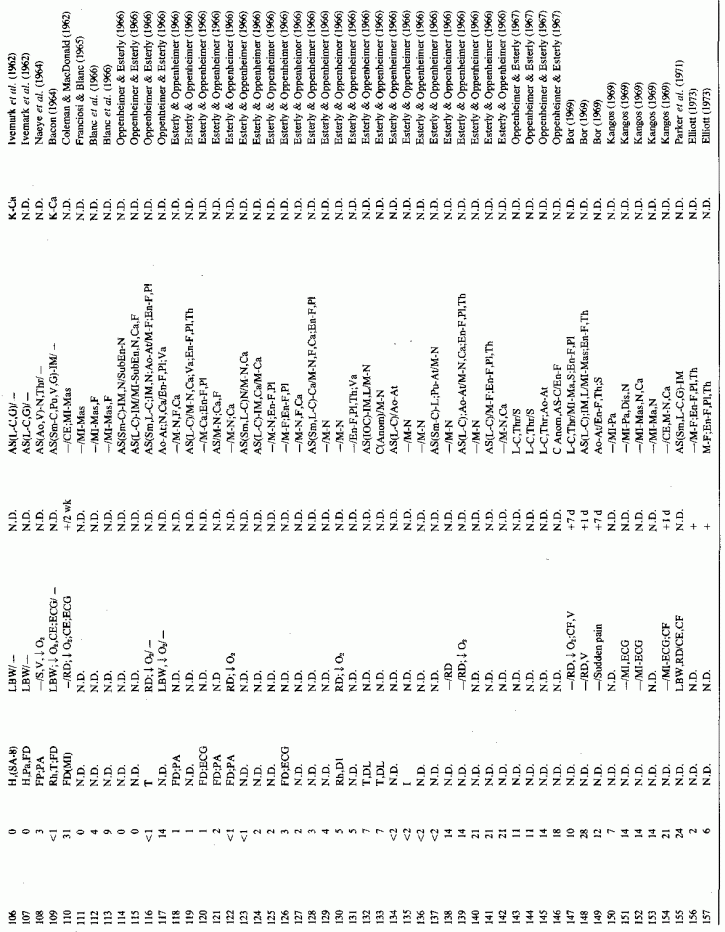
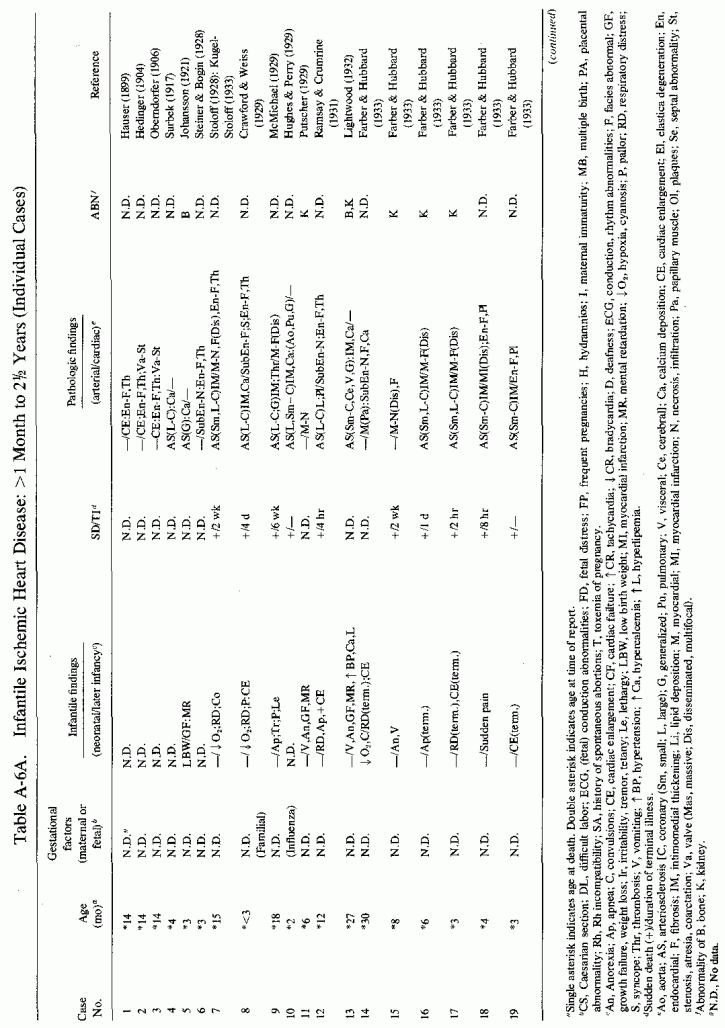
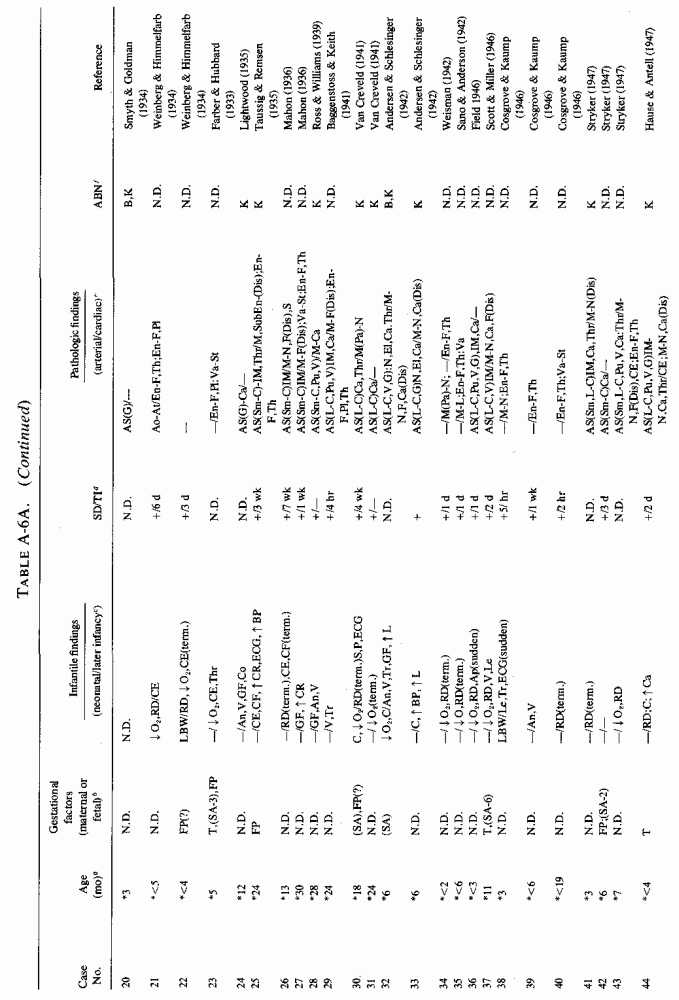
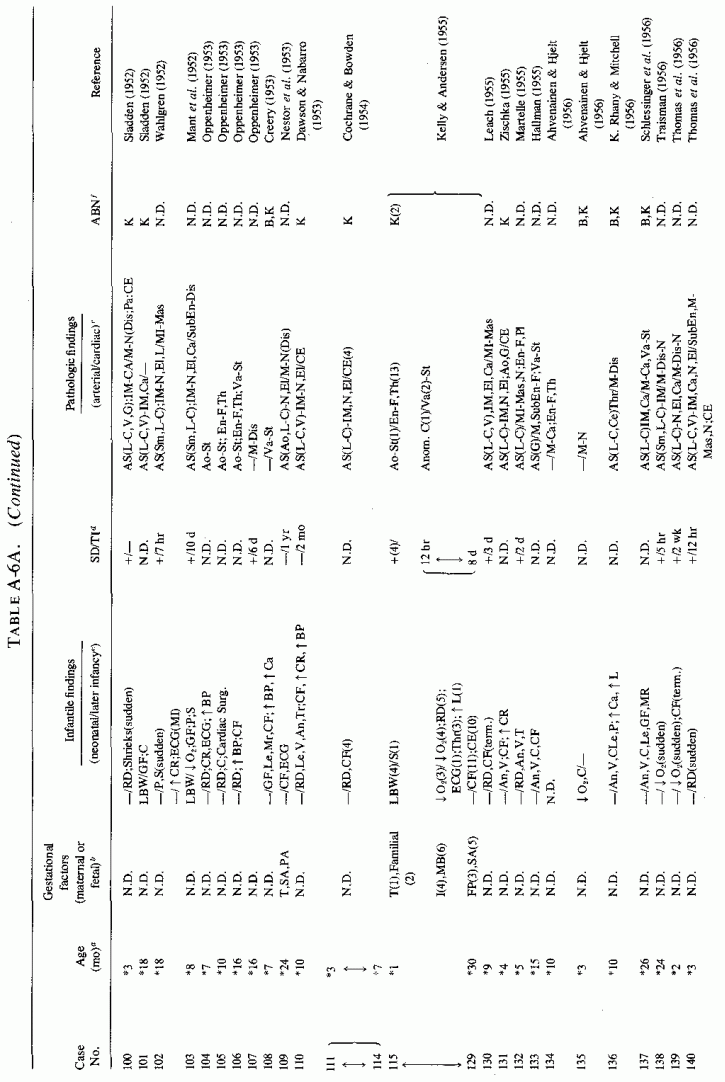
6.1.2. Pathology of Infantile Arteriosclerosis (See Appendix Table A-5A,Table A-5A continued, Table A-5A continued (2), Table A-5B, Table A-6A, Table A-6A continued, Table A-6A continued (2), Table A-6A continued (3), Table A-6A continued (4), Table A-6A continued (5),Table A-6A continued (6), Table A-6B and Table A-6b continued.)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Intimal, subintimal, and medial arterial lesions, usually of the small- and medium-sized arteries, such as have been described in infants who died suddenly or after protracted congenital cardiac disease, are characterized by elastica degenerative processes, mucopolysaccharide or calcium deposition, and proliferative or fibrotic intimal and medial changes. Lipid droplets are often also seen, but the fat deposition does not become atheromatous until later in infancy and childhood. The very early infantile arterial lesions resemble those of magnesium deficiency in animals with otherwise balanced diets, i.e., "pure" magnesium deficiency. Suddenly fatal arterial lesions of infants have usually been coronary (associated with perivascular myocardial microfocal necrosis, or more rarely with gross infarctions). However, most of the infants with coronary lesions also had arteriosclerosis of other viscera and occasionally had generalized arteriosclerosis. Whether earlier arterial lesions exist in infants who develop "adult-onset" atherosclerosis or in infants born to parents with early cardiovascular disease is difficult to ascertain.
Even among infants identified as having had cardiovascular disease pre- or postmortem, the degree and location of arterial damage are often not specified. Among the 157 separately cited cases of infants born dead or dying within the first month of life with cardiac lesions, 30 had coronary arteriosclerosis described and 14 had visceral or generalized arteriosclerosis described. Although not mentioned, coronary arterial lesions are probable in at least 80 more who had myocardial lesions ranging from necrosis with and without calcification to fibrosis. Only 5 of the 80 with endocardial fibroelastosis had coronary arterial lesions mentioned. Among the 253 infants tabulated as having died of cardiovascular disease from 1 month to 2 1/2 years of age, 85 were described as having coronary arteriosclerosis, with or without involvement of other arteries. Myocardial lesions suggestive of ischemic heart disease were described in an additional 74 infants, whose coronary arterial status was not described. Almost half of the 110 infants with endocardial fibroelastosis did not have coronary arterial or myocardial lesions described. Among those whose arterial lesions were described, a third of those up to 1 month of life had intimomedial proliferation and almost as many had thrombosis noted. About half of the infants of 1 month to 2 1/2 years of age, whose arteries were described, had intimomedial proliferation, but only a tenth had thromboses. It is not possible to ascertain the incidence of intimomedial proliferation from surveys of autopsy material, for some include intimal sites of proliferation, "cushions" as precursors of atheromata (Dock, 1946; Fangman and Hellwig, 1947), and others specifically exclude them as normal variants (Schornagel, 1956; Oppenheimer and Esterly, 1967).
With one exception, the 19 children whose arteries showed degenerative or calcific changes were no more than 4 days old at death. This might be supportive of Gruenwald's (1949) conclusion that perinatal hypoxia can cause arterial necrosis, based on his finding such lesions in as many as 9.5% of infants autopsied after stillbirth to 3 days of life. There were fewer instances of intimomedial degenerative changes in the older infants, but more instances of calcification. Three cases of lipid deposition in the arteries were noted in the individual case reports of infants up to one month of age; 6 were noted in the group up to 21/2 years.
Few patients with supra- or subvalvular aortic stenosis or with cardiofacial peculiarities are cited; most survived beyond the 2 1/2-year limit selected. That these children probably developed their abnormality either in utero or in the first 2 years of life seems likely.
This is a disease, the incidence of which is impossible to estimate. As a result of the effort to classify infants with histopathologically identical lesions as suffering from different diseases, depending on coexisting anomalies or demonstration of conditions that predispose to metastatic calcification, there is not uniformity of reporting. Further complicating the determination of the incidence of infantile coronary arteriosclerosis is the lack of agreement as to what the infantile arteriosclerotic lesion is. In "idiopathic" infantile arteriosclerosis, intimal thickening and elastica degeneration are recognized as the typical findings, but focal intimal proliferative, termed "cushions" (usually with fibromuscular disorganization of the media), which are found more than twice as often as are atheromas, are not uniformly considered pathological. When only atheromatous lesions are considered evidence of arterial disease, neonatal focal myocardial necrosis has been reported in the absence of lesions of the main coronary arteries. Rarely are the intramyocardial arteries examined. Thus, coronary occlusion or significant coronary disease is less frequently reported than is that of the myocardium or endocardium. Nonetheless, an attempt to select, from the pathology surveys, cases designated by the age groups selected here, and that exclude major anomalies (other than atresia of the great vessels), suggests about 500 in which hypoxia of the heart might have been involved in infants up to one month of age, and over 2000 in those from 1 month to 2 1/2 years of age.
In the case of endocardial fibroelastosis (EFE), myocardial ischemia has been repeatedly implicated. J. M. Craig (1949), who presented 43 cases, noted that microscopic myocardial necrosis and fibrosis was common. He suggested intramural coronary disease in utero as a contributory factor. F.R. Johnson (1952) suggested that intrauterine anoxia might be contributory to the EFE seen in malformed hearts; Moller et al. (1966) noted that infarcts of the papillary muscles are not infrequently found in infants with EFE. Since the subendocardial myocardium obtains oxygen from the blood in the heart chambers, conditions that interfere with blood outflow, that lead to stagnation, can lead to hypoxic subendocardial and endocardial damage and thickening. In fact, outflow obstruction is the most common anatomic disorder associated with EFE (Moller et al. 1964; Bryan and Oppenheimer, 1969).
A survey of necropsy material in a major medical center showed that myocardial infarction is not rare in infants, even occurring in utero (Franciosi and Blanc, 1968). In infants with congenital heart disease, the infarcts were limited to papillary muscles (which are supplied by the small end-arterial branches of the coronaries), and to microscopic lesions of the subendocardial ventricular myocardium that were adjacent to perivascular and interstitial fibroses. Although none was associated with occlusive arterial disease, grade 1 to 4 coronary lesions were found frequently. Grade I was characterized by frayed intimal elastica lamina; grade 2 additionally had slight focal intimal fibrosis; grade 3 had intimal cushions in addition; grade 4 had diffuse elastica fraying and diffuse intimal thickening equaling the thickness of the media. The frequency of the infantile myocardial infarcts was 80% among those with anomalous venous return, 89% in those with pulmonary valvular stenosis, and 100% in those with aortic valvular stenosis.
The coronaries are often not examined, even among infants who die during the perinatal period and are autopsied. This is particularly so in the case of the small- and medium-sized arteries, which are most often involved in infantile coronary arteriosclerosis, and which are most likely to be involved in focal and microscopic myocardial necrosis and fibrosis and in fibroelastosis. Blanc et al. (1966) pointed out that systematic examination of the small- to medium-sized coronaries of infants has disclosed that as many as 12% had arteriosclerosis.
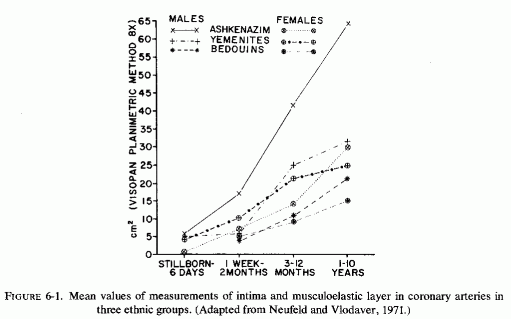
Despite the fact that many of the infants with necropsy evidence of coronary disease had died suddenly, none were recorded as having been reported by medical examiners or coroners (Moran and Becker, 1959). Thus, it seems likely that many of the instances of this disease are not recognized. Supporting the contention that many cases might be missed are the studies of autopsy material that include examination of the large coronaries of infants. In a study of the proximal segments of the main coronaries of 105 individuals who died before birth to the early twenties, only the fetuses (24 of 3½-9 months gestation) were free of coronary lesions (Moon, 1957). In that series, two premature infants had ruptured internal elastic membranes but had no other coronary lesions. Most of the 52 infants under two years of age had coronary lesions, the earliest noted being rupture and degeneration of the internal elastic membrane. Some also had fibroblastic proliferation with deposition of mucopolysaccharides and proliferation of endothelial cells overlying these areas. Infants several months old had progression of the intimal lesions as compared with newborn infants, the intimal thickening being very pronounced at three or four months of age. The intima was commonly thicker than the media. Serial sections of the left anterior descending coronaries of 88 infants, from stillborn to one year of age, also showed that intimal thickening increased with the infants' age (Schornagel, 1956). Grading the lesions I (endothelium on regular or split elastica interna) to III (thick intima), about 40% of the males had grades II and III lesions at less than one day to one month, and 24% and 37% of the females at less than one day and up to one month, respectively. Infant boys and girls of one month to one year had grades II and III coronary lesions in 91.3% and 87.5% respectively. That the earliest coronary lesions in the youngest infants is elastica degeneration, often without overlying intimal thickening, was attested to by Levene (1956), Gillman (1959), and Kaunitz (1961). The intimal hyperplasia, usually in the areas with elastica damage, was pointed out in the early studies of Dock (1946) and Fangman and Hellwig (1947), both of whom stressed the preponderance of intimal thickening in male neonates. Because these neonatal coronary lesions are so common, there is controversy as to whether they are the earliest arteriosclerotic lesions or merely adaptive phenomena (Review: Neufeld and Vlodaver, 1971). This group confirmed the greater degree of elastica degeneration and overlying intimal fibroblastic proliferation, as well as muscle degeneration in the media, in male than in female Jewish neonates of European derivation (Ashkenazim) but found far less sex difference in intimal thickening among Yemenite (Mideastern Jewish) and Bedouin infants(Fig. 6-1) (Neufeld and Vlodaver, 1968, 1971). Histologic examination of right and left coronaries from 211 consecutive hearts from fetuses, infants, and children up to ten years of age showed significantly higher intima/musculoelastica ratios among the Ashkenazi males than among Yemenite or Bedouin males (Neufeld and Vlodaver, 1968; Vlodaver et al. 1969). Since the infants with the greatest degree of intimal damage (Fig. 6-2) were from the ethnic group with the highest rate of adult ischemic heart disease, it was considered likely that the early coronary lesions were indeed the precursors of the later coronary atherosclerotic lesions (Neufeld and Vlodaver, 1971; Neufeld, 1974).
{kind=link}
{kind=link}
Although coronary and myocardial lesions were most often the causative factors in the terminal event, most of the babies with coronary disease also had arteriosclerosis of other arteries, generally (in order of frequency) of the kidneys, adrenal glands, pancreas, spleen, lung, mesentery, and thyroid (Review: Moran and Becker, 1959).
The type of coronary arteriosclerosis, particularly of the small-to-medium coronary arteries, and the perivascular focal myocardial necrosis (that are seen in infancy) strongly resemble the coronary and myocardial lesions produced in animals on magnesium-deficient diets (Seelig and Haddy, 1976/1980). Most of the magnesium-deficient animals with cardiovascular lesions were immature. There have been comparable changes reported in herbivores however, usually during early lactation, occurring in herds grazing on magnesium-poor lands, on pasturage with factors interfering with availability of magnesium, and not infrequently in herds with a high incidence of eclampsia (Arnold and Fincham, 1950; Lynd et al., 1965; Willers et al., 1965; review: Seelig and Bunce, 1972). Data on abnormalities during pregnancy or delivery are not frequently given in papers on infantile cardiovascular disease. Some information was given in a third of the tabulated infants who died under one month of age and in less than a third of those with the disease reported in infants from one month to two and a half years of age (Appendix Table A.6A). Abnormal or frequent pregnancies, long or complicated deliveries, immaturity during gestation, multiple births, and maternal diabetes mellitus-all conditions that have been associated with low levels of magnesium-and placental insufficiency or premature separation of the placenta are conditions associated with prenatal hypoxia and malnutrition. Several of these factors were cited in eight of 29 infants dying with myocardial lesions, who had fetal distress recorded (Oppenheimer and Esterly, 1967). Such factors plus hydramnios or RH incompatibility were reported in 63 of the 157 of those under one month of age (Appendix Table A-5A) [and (Appendix Table A-5A continued)], but in only 38 of the 251 infants over one month to two and a half years old (Appendix Table A-6A) [and (Appendix Table A-6A continued),(Appendix Table A-6A continued (2), (Appendix Table A-6A continued (3), (Appendix Table A-6A continued (4), (Appendix Table A-6A continued (5), and (Appendix Table A-6A continued (6), few of whom had maternal histories cited. There were several instances in which there had been previous unsuccessful pregnancies, or in which siblings or close relatives had died similarly. Thus, it seems that metabolic disorders or gestational stress (especially in instances of maternal immaturity, or frequent or multiple pregnancies) might have played roles in absolute or conditioned magnesium deficiency. Unfortunately, magnesium levels were almost never recorded in the propositus or mother, leaving speculative the supposition that magnesium deficiency might have been contributory in the cited cases. An exception is the infant, reported by Vainsel et al. (1970), who had hypomagnesemic hypocalcemia and whose refractoriness to vitamin D and calcium therapy appeared to be familial. He and three male siblings (out of six) had had convulsive seizures. One died at six weeks; the described infant died at three months and was found to have focal myocardial necrosis and coronary calcinosis. Since he was the ninth infant in his family, both a metabolic and multiparity-induced hypomagnesemia might have participated in his severe hypomagnesemia (0.4-0.65 mEq/liter), the magnesium deficiency having been detected only a few days before death (Vainsel et al., 1970). Until prospective and retrospective magnesium data are obtained from affected infants and their mothers, from subsequent pregnancies and infants, and from near relatives, the validity of the premise that magnesium deficiency is contributory to infantile arteriosclerosis and its complications remains untested.
The medial necrosis of the coronaries seen in large infants with birth asphyxia (Gruenwald, 1949) might also be related to loss of tissue magnesium. Perhaps sufficient magnesium can leave the tissues of the coronary arteries and the heart to cause necrosis or arrhythmia or both. The intimal and medial loss of functional myocardial magnesium (Review: Seelig, 1972) might participate in the cardiac lesions of infantile cardiovascular disease.
Perhaps contributing to infantile coronary arterial lesions and microfocal myocardial necrosis (that either results in immediate death or sets the stage for cardiac death in the later months or years) is neonatal hypoparathyroidism. Lehr and his colleagues (Lehr, 1965, 1966; Lehr et al. 1966) have shown that parathyroidectomized rats, particularly when they are phosphate loaded, develop lesions of the small coronary arteries and perivascular microfocal myocardial necrosis. The high phosphate content of cows' milk, fed to infants during the neonatal period, especially when their parathyroid hormone secretion is often subnormal, and the hyperplasia of infants' coronaries might also be related to episodes of hypoxia, conceivably such as are experienced by infants that suffer from periods of sleep apnea (as in the SIDS). The production of severe arteriosclerosis, predominantly of the arterial connective tissues, by exposure of rabbits to short periods of hypoxia daily for two weeks (Helm et al., 1969; Garbarsch et al., 1969) would seem to support that supposition. Not noted in Gruenwald's (1949) study of large infants whose medial necrosis was attributed to perinatal anoxia was whether any had been born to diabetic mothers. Infants of diabetic mothers not only tend to be large but have also been found to have a high incidence of hypomagnesemia.
In the infants past the neonatal period, the use of cows' milk formulas that not only provide a substantial phosphate load, but that also provide vitamin D additional to that generally prescribed by the physician, can also contribute to magnesium deficiency. The generalized arteriosclerosis, valvular disease, and fibroelastosis of babies that have received excessive vitamin D or that are hyperreactive to it have been reviewed (Seelig, 1969b, 1978; Seelig and Haddy, 1976/1980; Seelig and Mazlen, 1977). Regarding its effects during pregnancy, experimental hypervitaminosis D has been implicated in placental abnormalities, such as those that contribute to fetal malnutrition, anoxia, and possibly to eclampsia. It is well to remember, thus, that vitamin D excess causes magnesium loss that might well be implicated in infantile cardiovascular disease (Seelig and Haddy, 1976/1980).
Since most of the studies of the pathogenesis of atherosclerosis have focused on fat, and most studies of magnesium deficiency were with animals (usually rats) whose control and experimental diets were also high in calcium, phosphate, and often vitamin D (Reviews: Larvor and Durlach, 197 lb; Seelig and Haddy, 1976/ 1980), there are few experimental studies of the vascular changes caused by magnesium deficiency alone (Fig. 6-3). Lowenhaupt et al. (1950) reported that young rats kept on a normal diet, except for magnesium deficiency, developed myocardial lesions (within two weeks) around the small coronary radicals of precapillary and capillary size. Other magnesium-deficiency studies, that elicited focal myocardial infiltration, necrosis, and scarring (Mishra, 1960a; Mishra and Herman, 1960; Seta et al., 1965) are suggestive of damage to the small intramyocardial coronaries. Heggtveit (1965c) reported edema of the small coronary arteries in Mg-deficient rats. Hungerford and Bernick (1976/1980) have provided details of the nature of the coronary arterial damage produced by magnesium deficiency in rats: intimal thickening with extracellular edema, thinning of the internal elastic membrane with disruption, and disorientation and hyperplasia of medial muscle cells. Some of the arteries had densely aggregated pyknotic cells in their enlarged tunica media, with narrowed lumina. This group confirmed the inflammatory changes of the perivascular myocardium, reported more than 25 years earlier by Lowenhaupt et al. (1950). Dogs on otherwise balanced magnesium-deficient diets had pyknotic intimal cells in small coronary arteries and arterioles, but no intimal hyperplasia; their medial muscle cells were loosely arranged, suggestive of edema, with necrosis and inflammation. The larger coronaries were less damaged (Wener et al., 1964). Intimal and medial calcification were described in magnesium-deficient dogs, despite decreased serum calcium levels (Syllm-Rapoport and Strassburger, 1958; Unglaub et al., 1959; Bunce et al., 1962b; Featherston et al.1963., Morris et al., 1963; Wener et al., 1964). As in the magnesium-deficient rats, which had perivascular myocardial necrosis and edema (supra vide), magnesium-deficient dogs had comparable myocardial lesions (Unglaub et al.,1959; Wener et al., 1964). Large myocardial infarcts were seen in severely magnesium-deficient dogs (Morris et al. 1963).
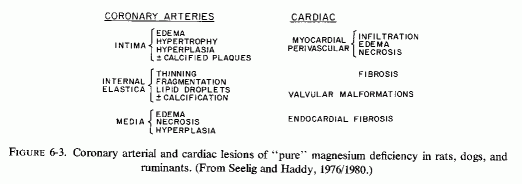{kind=link}
The histologic arterial changes of magnesium deficiency were first characterized in cows, on spring forage on magnesium-poor soil, or where other factors interfered with the availability of magnesium (Arnold and Fincham, 1950). These observations were reaffirmed in controlled studies (Lynd et al.1965; Willers et al. 1965). The coronary arteries and the endocardium showed intimal thickening and subendothelial degeneration and calcification of elastica fibers. Medical calcification and calcific intimal plaques were also described, as were valvular malformations and myocardial infarcts (Willerset al., 1965). The syndrome, leading to these cardiovascular lesions, was seen predominantly in lactating cows in areas where "grass tetany" or convulsions of magnesium deficiency (characterized by hypomagnesemia and hypocalcemia) occurred during late pregnancy or during lactation. Not only cows but ewes are susceptible to this disorder, and it has been noted that it is more prevalent in herds with a high incidence of toxemia of pregnancy (Herd, 1966a,b).
6.2.1.2. Arterial Damage of Magnesium Deficiency, Intensified by High Calcium and Vitamin D Intakes (Fig. 6-4)
{kind=link}
High dietary calcium/magnesium dietary ratios have uniformly increased the susceptibility to the symptoms and signs of magnesium deficiency. It is important to remember that high intakes of calcium interfere with magnesium intestinal absorption and increase its renal excretion, and that high intakes of vitamin D also favor calcium retention over that of magnesium (Reviews: Seelig, 1964, 1971).
Cardiovascular changes, similar to those seen in "pure" magnesium deficiency, developed in rats fed 400-650 times as much calcium as magnesium (versus 40/1 in controls, which is also a much higher than normal Ca ratio). There were small inflammatory and necrotic myocardial lesions (suggestive of disease of the small coronary arteries) with increased tissue calcium and sodium, but no significant change in serum calcium, and low tissue and serum magnesium and potassium (Mishra, 1960a; Ko et al., 1962). Adding sufficient magnesium to lower the Ca/Mg ratio to 3/1 prevented the lesions (Mishra, 1960a). In a study designed to show how much magnesium is necessary to prevent macroscopically manifest intimal calcific plaques in dogs on Ca/Mg intakes of 33-50/1, Bunce et al. (1962a) found a little more than a twofold increase in grossly visible aortic intimal lesions in dogs receiving 0.6% of calcium (Ca/Mg = 33/1). Most of the dogs on high calcium/low magnesium intakes had intimal plaques.
Since vitamin D normally increases serum calcium levels and increases magnesium requirements, it is of interest that magnesium-deficient dogs on normal calcium intakes showed minimal coronary arterial calcification unless they were given vitamin D or an intravenous calcium load (Syllm-Rapoport and Strassburger, 1968; Unglaub et al., 1959). An early study (Handovsky and Goormaghtigh, 1935) showed that moderately high doses of vitamin D significantly raised the blood pressure in dogs; that vitamin D excess causes arteriosclerosis has been known even longer (Kreitmair and Moll, 1928). Like the arterial lesions of magnesium deficiency, those of experimental hypervitaminosis D (Gillman and Gilbert, 1956) involve medial and elastica degeneration and calcification (Review: Seelig, 1969), but the predominant lesions described were of the larger arteries, rather than of the coronaries. Rats on toxic doses of vitamin D also developed hypercholesterolemia, hypertension, and aortic calcification; the latter changes were prevented by high-dosage magnesium supplementation (Sos et al., 1960; Rigo, 1965; Rigo et al., 1965a; Sos, 1965).
When calves were fed low-magnesium diets that were usually comprised of whole milk or a comparable synthetic diet (both supplemented with vitamin D) for prolonged periods, they developed neuromuscular signs of magnesium deficiency and endocardial and intimal plaques, and fragmentation, degeneration, and calcification of the elastic fibers of both endocardium and arteries, and phlebothrombosis and focal myocardial necrosis (Moore et al., 1936, 1938; Blaxter et al., 1954). The magnesium-deficiency syndrome was prevented by magnesium supplementation: 30-40 mg/kg/day (Huffman et al., 1930; Duncan et al., 1935; Moore et al., 1936, 1938; Blaxter et al., 1954). Like human infants, despite these calves' high calcium intakes, their serum calcium remained normal or slightly low. In addition to the endocardial and intimal calcification, calves on a whole-milk diet developed high serum cholesterol levels (J. W. Thomas, 1959).
{kind=link}
Studies of the influence of magnesium deficiency and its repletion on the development of atherosclerosis in rats (fed different combinations of saturated or unsaturated fat diets with and without added cholesterol and cholic acid and calcium) (pages 148-152) have shown dissociation between serum and cardiovascular lipids. It is noteworthy that increased magnesium intakes of animals on atherogenic, hyperlipidemic diets decreased arterial and myocardial lipid deposition without lowering the elevated serum lipids; the magnesium even raised the serum lipids somewhat (Vitale et al., 1957d; 1959; Nakamura et al., 1960). In contrast, high calcium intakes lowered the serum lipids but raised the arterial lipids (Vitale et al., 1957c; 1959; Hellerstein et al., 1957, 1960; Nakamura et al. 1960). Long-term administration of magnesium to rats on atherogenic diets, which only gradually lowered serum lipids to a minor degree, resulted in more rapid and significantly reduced arterial lipid deposition (Nakamura et al., 1960, 1966). The rats on the low-magnesium, high-fat diets were the only high-fat-fed rats to develop fat deposition in heart valves and plaque formation in the aorta (Nakamura et al., 1966). In this series of experiments, subintimal and medial degeneration and calcification of the elastica, as well as intimal atheromata, developed only in the rats that were magnesium deficient as well as fat loaded. Calcification of the media of pulmonary artery and of the myocardium (some with interstitial inflammatory infiltration) were also noted in magnesium-deficient, fat-loaded rats. In a further study to explore the mechanism of the intensification of atheroma formation by magnesium deficiency of rabbits on an atherogenic diet, Hirano (1966) measured the uptake of radioisotope "C-tagged cholesterol by the heart, aorta, and other viscera. Rabbits fed the magnesium-deficient atherogenic diet showed increased radioactivity in the aorta, as compared with controls. Even magnesium-deficient rabbits on low-cholesterol intakes had increased fat deposition in the aortas, but to a lesser degree. Despite the increase in aorta cholesterol in magnesium-deficient rabbits, the serum cholesterol level was not significantly altered.
Nakamura et al. (1965) found that rabbits that had developed atherosclerosis required substantial amounts of magnesium added to their diets to exert a notable effect on atherogenesis. More than 950 mg/100 g of diet was necessary to affect serum and tissue lipids. The authors commented that aortic lipid deposition is significantly enhanced by magnesium deficiency; high magnesium intake merely slows the process. The elevated intimal plaques, fragmented and calcified elastica, and mural thrombi that were reported in the magnesium-deficient rabbits, were not seen in the matched cholesterol-loaded, magnesium-supplemented rabbits; they showed no calcified lesions and less foam cells in the subintimal layer of the aorta (Nakamura et al., 1965). Narrowing of coronary arteries was noted in all of the cholesterol-loaded rabbits, but to a somewhat lesser degree in the rabbits on high magnesium intakes. Greater involvement of the small coronary arteries is suggested by the microscopic foci of myocardial necrosis in half the rabbits on magnesium-deficient, high-cholesterol diets, but in none of those that were magnesium supplemented. Bunce et al. (1962a) showed that increasing the magnesium intake, sufficiently to prevent intimal lesions in dogs on a high saturated fat diet, actually increased their serum cholesterol levels, whereas the dogs on the highest Ca/Mg ratio had lower serum cholesterol (270%) and more intimal lesions. Dogs on a magnesium-free, corn-oil-rich, low-calcium diet had intimal thickening and plaques with narrowed coronary lumens, but minimal lipid deposition (Vitale et al., 1961). Monkeys on a similar diet exhibited raised intimal atheromata and fibroblastic intimal thickening, with disrupted elastica, but no arterial calcification.
These findings raise the question as to whether seeking to correlate serum magnesium and cholesterol levels provides meaningful data regarding the influence of magnesium deficiency or therapy on atherosclerosis. Even when high doses of magnesium are given to hypercholesterolemic animals, the changes in serum lipids are less consistent than is the lowering of tissue lipids. The serum levels of cholesterol have been unaffected, or even raised in some of the studies; the β-lipoprotein fraction seems to be influenced somewhat more. Although magnesium deficit intensifies atheromatosis, it takes quite large doses and/or prolonged administration of magnesium to protect against the disease in hyperlipemic animals (Hellerstein et al., 1957; Rigo et al., 1963, 1965a,b; Nakamura et al., 1965, 1966). It seems that neither the serum magnesium nor cholesterol level are illustrative of the tissue levels. Thus, to determine the effect of magnesium on lipids in man, we must investigate the response to effective doses of magnesium. The preliminary clinical trials cited in this chapter are not conclusive. Prolonged trials with more intensive exploration of the leads mentioned here are indicated. The effect of magnesium on high-density lipids needs study.
6.2.1.4. The Cardiovasopathic (CVP) Diet
An experimental diet (Table 6-2) has been devised that causes spontaneous myocardial infarctions (MI) in 80-90% of the animals (rats, dogs, and cocks) fed that diet but kept under otherwise normal conditions (Sos et al., 1960, 1964a,b,c; Sos, 1965; Rigo et al.,1961, 1963a,b, 1965a,b; Rigo, 1971; Gati et al., 1964, 1965; Szelenyi, 1971, 1973; and Review: Seelig and Haddy, 1976/1980). With the exception of low chloride, it possesses the characteristics of diets consumed by many in our affluent society. It is high in fat, cholesterol, vitamin D, sodium, phosphate, and protein; it is low in magnesium, potassium, and chloride. In addition to the massive infarctions, animals on the CVP diet had atherosclerosis, hyperlipidemia, and the abnormalities (Table 6-3). Without the added cholesterol, animals on the (modified) CVP diet still had high cholesterol levels, but they were half as high as those on the complete CVP diet. The hypertension was unaffected, but the incidence of MI dropped to 60% of the group. Elimination of only vitamin D did not lower the blood cholesterol, but the animals had only slight hypertension, and fewer (40%) developed MI. Halving the protein content of the CVP diet (to a normal intake) resulted in a slight increase in serum cholesterol, no change in the hypertensive level, but resulted in about half the MI incidence (40%) of the CVP animals. Providing a normal salt mixture lowered the cholesterol somewhat but not the hypertension. It lowered the incidence of infarction to 13%. Increasing the dietary intake of magnesium chloride fivefold over the normal requirement mitigated, significantly, the cardiopathic changes as well as the coronary and aortic pathology, which had included thickening of the small coronary arteries, with marked increase of the arterial wall/lumen ratio (Sos, 1965; Szelenyi, 1971). The increased magnesium intake also reduced the extent of damage produced by such intensifying factors (added to the CVP diet) as neurogenic stress, or ACTH. When the CVP diet was modified by increasing the cholesterol threefold, the fat fourfold, vitamin D and cod liver oil 1/3 each, and adding thiouracil, marked hypercoagulability was produced. Fivefold increased magnesium intake restored the coagulation and prothrombin times to normal (Szelenyi, 1971, 1973).
{kind=link}
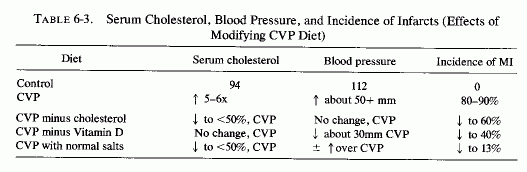{kind=link}
The rats on the CVP diet retained 15 times as much sodium as did the controls, but their myocardial and serum sodium levels differed little from control values. Their myocardial calcium rose 12%, but their serum calcium remained essentially unchanged. Their myocardial magnesium and potassium levels dropped 19 and 33%, respectively; serum values of both cations dropped about 20% (Sos, 1965; Table 6-4).
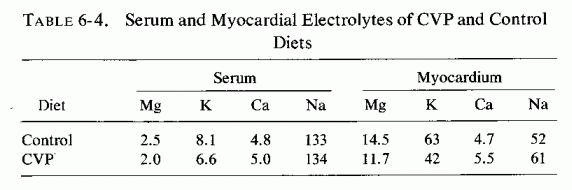{kind=link}
Arterial lesions, similar to those produced by magnesium deficiency, in combination with high calcium, vitamin D, or fat intakes or other imbalances (i.e., CVP diet) have been produced by modalities that increase serum calcium or cholesterol levels, increase tissue sodium and calcium levels, and decrease magnesium and potassium, both in the serum and in the tissues. (Table 6-5). Dihydrotachysterol, particularly in combination with sodium acid phosphate (NaH2PO4) causes coronary and aortic calcification and periarteritis, lesions that are intensified by magnesium or potassium deficiency, partially protected against by administration of either cation and better protected against by both and by the chloride ion. (Selye, 1958a,b; Bajusz and Selye, 1959; Mishra, 1960d). Mineralocorticoids plus phosphates produce multifocal necrosis (suggestive of small coronary disease), the intensity of which is also increased by magnesium and/or potassium deficiency; again, each cation is protective (Selye, 1958a,d,e,f; Selye and Mishra, 1958; Bajusz and Selye, 1959; Mishra, 1960b; Selye and Gabbiani, 1965). Parathyroid extract, with sodium phosphate salts (Selye, 1958c; Lehr, 1963) or stimulation of parathyroid secretion and/or adrenal medullary and cortical secretion, as occurs in renal damage or nephrectomy (Lehr, 1959), causes subintimal arterial damage with calcification of the damaged elastica, in addition to myocardial infiltration and edema. Administration of mineralocorticosteroids markedly intensifies the cardiovascular lesions of these (Lehr, 1959) and of the catecholamine myocardial necrosis model (Guideri et al., 1971). Paradoxically, despite the calcium-mobilizing effect of parathyroid hormone, and the vitamin-D-like arterial damage it produces in combination with a phosphate salt, Lehr (1959) has shown that phosphate-loading of parathyroidectomized rats causes even more severe cardiovascular lesions. Subsequent work from his laboratories has demonstrated that the common denominator in the experimental models-calcium- or phosphate-loading in the presence or absence of parathyroid hormone, or with mineralocorticoid, or catecholamine (exogenous or endogenous)-is depletion of myocardial magnesium and subsequently of potassium (Lehr, 1965b, 1969; Lehr et al., 1966, 1969, 1970/1972, 1976/1980). The increase of cellular calcium reflects, predominantly, the calcification of injured tissues, even in the presence of hypocalcemia of the parathyroidectomized rats. Stress has also been associated with markedly increased myocardial damage when the animals are magnesium or potassium deficient, and magnesium administration has protected against stress and exogenous catecholamine-induced cardiovascular damage (Selye, 1958g; Selye and Mishra, 1958; Shimamoto et al., 1959; Mishra, 1960e; Mishra and Herman, 1960; Bajusz, 1965a). Lehr (1965, 1966) has correlated the microfocal myocardial necrosis, seen in most of the drug- and stress-related experimental cardiovasopathic models (which resemble the lesions of "pure" magnesium deficiency,supra vide), with damage to the cardiac microcirculation, with medial degeneration and perivascular myocardial necrosis, and has stressed the depletion of intracellular magnesium as an early and consistent change. The animals that are loaded with calcium, vitamin D, and/or fat: all agents that cause hypercholesterolemia, hypertension, or thrombogenesis seem to have a greater tendency to develop infarcts (supra vide: CVP diet). That magnesium deficiency predisposes to the hypercoagulability, and that magnesium administration has been protective, may relate to the role of magnesium in platelet function (Review: Elin, 1976/1980), as well as to the effects of magnesium on coagulation factors (Szelenyi et al., 1967; Szelenyi, 1971, 1973; Stevenson and Yoder, 1972; Seelig and Heggtveit, 1974).
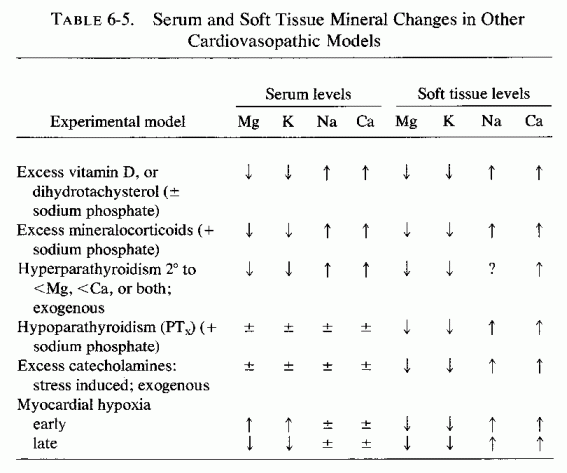{kind=link}
In addition to the increased susceptibility to atherogenesis that catecholamines can cause by inducing lipolysis, Raab (1958) called attention to the evidence that prolonged administration of small doses of epinephrine produces intimal thickening of small and large vessels of rabbits and dogs, and that larger doses produce necrotizing and calcifying lesions of the media. The similarity of these arterial lesions to those of magnesium deficiency, particularly in association with high intakes of calcium or of calcemic agents (supra vide), brings attention to the evidence that catecholamines cause loss of cellular magnesium. Epinephrine has been shown to increase plasma magnesium levels after its injection or after drug- or stress-induced stimulation of its secretion (Rogers and Mahan, 1959a,b; Larvor, 1968; Larvor and Rayssiguier, 1971; Rayssiguier and Larvor, 1971/1973). Catecholamine injection or its stress-induced secretion has caused lowered myocardial magnesium levels. This effect might be partially reciprocal to catecholamine-induced cellular uptake of calcium, a physiologic action that contributes to its positive inotropic effect (Nayler, 1967). The arterial damage caused by catecholamines, however, must be a pathologic extension of its activity that intensifies production of a low cellular magnesium/calcium ratio.
One mechanism might be via local hypoxia mediated by proliferative constrictive endothelial proliferation, in conjunction with its increase of oxygen consumption (Raab, 1969). It should be recalled that even short-term local hypoxia, such as is produced by occluding the vessels by a blood pressure cuff can cause increased plasma magnesium, presumably as a result of egress of cellular magnesium (Whang and Wagner, 1966; S. P. Nielsen, 1969). Thus, this mechanism, too, can produce a low Mg cellular ratio.
The general increase in blood pressure that is the classic response to catecholamine release or injection must also be considered. The cardiac output increases as a result of increased strength of myocardial contraction and increased heart rate [both contributed to by the catecholamine-stimulated shift of calcium into the heart (Nayler, 1967)], and secondary to the increased venous return to the heart, as splanchnic, renal, skin, and mucosal arterioles constrict.
The protection by magnesium against intimal damage (supra vide) might serve to protect the arterial lining from the mechanical stresses caused by sudden changes in pressure and local oxygenation. There are experimental data suggesting that magnesium deficiency increases some of the catecholamine effects on the arteries, and that magnesium excess tends to counteract them. For example, Hanenson (1963) found that absence of magnesium from the medium in which aortic slices were suspended markedly increased the contractile response to norepinephrine; its addition decreased the contractile response. However, recent work on interrelationships of magnesium and calcium with vasoactive hormones on vascular muscle has elucidated the magnesium dependence of the reactions and explained how magnesium depletion can cause refractoriness to vasoactive hormones (pages 179-183). In vivo rat studies have shown that slow intravenous infusion of magnesium sulfate decreases the hypertensive response to epinephrine or norepinephrine (Cession et al., 1963). The influence of magnesium deficiency or excess on release of catecholamines is considered under our discussion of magnesium and the heart. In this regard, the demonstration that arterial tissue exhibits rapid uptake of catecholamines (particularly of epinephrine) even when injected within the range that is probably produced by catecholamine-releasing agents, such as nicotine, stress, hypoglycemia, or thyroid hormone (Raab and Gigee, 1958) is probably relevant to the clinical situation.
A high-fat diet that was thrombogenic (incorporating propylthiouracil and cottonseed oil) became cardiopathic when NaH2PO4) was added (Savoie, 1972a,b, 1975). Since the findings with this regimen, and with modifications of the electrolyte steroid cardiac necrosis (ESCN) syndrome developed by Selye (supra vide), including catecholamines and exposure to stress, point predominantly to effects of drugs and minerals (predominantly magnesium) on cardiac metabolism (Savoie, 1971a,b, 1975) infra vide.
Still another change has been seen in the cardiovascular tissues of both atherosclerotic experimental animals and patients, and in experimental magnesium deficiency: decreased numbers of mast cells and evidence of degranulation. It has been suggested that connective tissue mast cells may play a role in the development of arteriosclerosis (Constantinides, 1953; Cairns and Constantinides, 1954; Wexler (1964). Evidence for this theory derives from the observation that rats, a species resistant to atherosclerosis, has many mast cells in the myocardium, whereas susceptible rabbits (Constantinides, 1953) and chickens (Padawer, 1957) have few mast cells. Furthermore, young women have more mast cells than do young men and atherosclerotic patients have fewer mast cells than do normals (Hellstrom and Holmgren, 1950; Constantinides and Cairns, 1954). Wexler (1964) has reported that the number of myocardial mast cells was most severely depressed in breeder rats that developed the most severe spontaneous arteriosclerosis, and that mast cells were not found in the vicinity of the arteriosclerotic lesions. Their granules showed a marked change from metachromasia to orthochromasia, which was interpreted as indicating secretory discharge. The decrease in numbers of mast cells in arteriosclerotic arteries has been correlated with the decrease in hyaluronic acid in arteriosclerotic arteries (K. Meyer et al., 1959; Kaplan and Meyer, 1960; Buddecke, 1962). Since mast cells secrete hyaluronic acid (Padawer, 1957), the decrease in number of mast cells in arteriosclerosis may explain the decline in hyaluronic acid content. Experimental magnesium deficiency has also been shown to cause decreased tissue mast cells, and to increase their degranulation (Belanger et al., 1957, Bois et al., 1960; Hungerford, 1964; Hungerford and Bernick, 1976/1980). More data are required to ascertain whether the decreased numbers of mast cells in animals and patients with atherosclerosis might be contributed to by magnesium deficiency.
The term "hypertensive-arteriosclerotic cardiovascular disease" reflects the frequent association of what are actually two separate diseases: hypertension and arterio-, or more commonly, atherosclerosis. That hypertension can lead to arterial damage has been accepted for many years, although the mechanisms are still subject to dispute. This is not the place to consider the experimental evidence and hypotheses that suggest that hypertension can predispose to formation of atherosclerotic by damaging the endothelium and permitting diffusion of cholesterol into the intima (Haust, 1970), e.g., at sites of swirling and eddying of the blood stream (Duff, 1951), as a result of pressure-induced arterial dilatation (Schornagel, 1956; Helm et al., 1971). It is, however, pertinent that each disorder has been found in magnesium-deficient models, most commonly in combination.
The mechanism of the morphological changes produced in the blood vessels by magnesium deficiency is not clear, but the changes almost certainly contribute to the increased arterial resistance. A contribution by vasoconstriction also seems likely, particularly since magnesium deficiency (experimental) is usually associated with decreased serum concentrations of magnesium, potassium, and in soft tissues, decreased content of magnesium and potassium and increased content of calcium and sodium. Depending on whether the magnesium deficiency is associated with intake of calcemic agents, the serum calcium can be either low, normal, or high. When it is high, the electrolyte imbalance is one that has been shown to increase arterial resistance (infra vide). Increased plasma renin activity, blood serotonin level, and urinary aldosterone excretion have also been noted in magnesium deficiency-all factors that also increase arterial resistance.
The effect of magnesium deficiency on blood pressure involves complex interactions. Although most of the experimental models are associated with increased blood pressure, there are both clinical and experimental circumstances in which no effect, or actual lowering of blood pressure has been seen with magnesium deficiency. Insight into these paradoxical findings derives both from in vivomagnesium-deficiency studies and from in vitro investigations that have elucidated several aspects of the response of the vascular smooth muscle contractility and resistance to changes in magnesium and calcium concentrations.
Among the experimental models of cardiovascular disease are several that are characterized by decreased magnesium levels, and that are associated with increased arterial resistance. The magnesium-deficient animals that develop hypertension, as well as arterial and cardiac damage, usually also have hyperlipidemia, atheromata, and hypercalcemia. Animals with vitamin D toxicity, or those given the CVP diet (supra vide), fall into this category. Only in one study, in which the relative dietary intakes of magnesium and calcium were changed, did the blood pressure of calcium-deficient, magnesium-adequate rats show a greater decline than did that of magnesium-deficient rats (Itokawa et al., 1974b).
Acute studies, in which a low ratio of magnesium, potassium, or both, to calcium is produced by intra-arterial infusions, have shown that such a ratio (particularly in the presence of alkalosis) increases arterial resistance in peripheral, renal, and coronary circulation (Haddy, 1960, 1962; Haddyet al., 1963, 1969; J. Scott et al., 1961, 1968; Frohlich et al., 1962). On the basis of the experimental findings, Haddy and Overbeck (1962) and Frohlich et al. (1964) suggested that such electrolyte imbalances might be a common denominator in clinical hypertension. They cited hypercorticism, hyperparathyroidism, vitamin D toxicity, and eclampsia as diseases characterized by hypertension and often by such electrolyte abnormalities. Similar acute intraarterial infusion studies with normotensive and hypertensive human subjects yielded comparable results (Overbeck et al., 1969). The most recent reviews consider the physicochemical interrelationships among the cations and the arterial contractile processes (Altura and Altura, 1977a,b; Haddy and Seelig, 1976/1980). Calcium plays a central role in excitation-contraction coupling and this ion competes with magnesium for binding sites on the membrane of the vascular smooth muscle cell. When extracellular magnesium falls, more calcium is available on the membrane for entrance into the cell with each spike potential. Furthermore, the number of spike potentials may increase because lack of magnesium and potassium suppresses the sodium-potassium pump, which, because of the electrogenic nature of the pump, leads to a decrease in the resting membrane potential. Increased membrane calcium and number of spikes would elevate intracellular calcium and cause vasoconstriction.
The magnesium concentration of the medium in which arterial strips are suspended affects their contractile response to vasoactive drugs and hormones. In its absence, potent contractile responses have been produced (Altura, 1970, 1975a,b; Altura and Altura, 1971, 1974, 1976/1980, 1978; Alturaet al., 1976/1980), an action that might be attributable to the greater surface binding of calcium when there is no competition by magnesium for common binding sites (Altura and Altura, 1971, 1974; Turlapaty and Carrier, 1973; Jurevics and Carrier, 1973). Somlyo et al. (1972) have proposed that incubation of arterial strips in magnesium-free solutions reversibly blocks the hyperpolarizing effect of cyclic AMP. Altura and Altura (1977, 1978) have hypothesized that these acute effects of changes in extracellular ionic magnesium result from effects on calcium permeability, translocation, and membrane stability, as well as from competition for binding sites. In fact, as with other tissues, exposure of vascular smooth muscle to low extracellular magnesium concentrations has resulted in increased tissue calcium content (Altura and Altura, 1971; Palaty, 1971, 1974). It can be speculated that reciprocal changes in serum and tissue calcium seen in magnesium-deficient animals (Review: Seelig and Haddy, 1976/1980) might be mediated by these mechanisms. Altura and Altura (1974) have provided evidence from their in vitro studies that withdrawal of ionic magnesium from the suspending medium produces arterial muscle contraction that is due to the inward movement of calcium. When a chelating agent (Ca EDTA) that selectively removes magnesium is added to the medium, the arterial muscle contracts; when one that selectively chelates calcium (EGTA) is added, the arterial muscle relaxes (Altura and Altura, 1975). Carrier et al. (1976) have shown that magnesium has two components in its interaction with calcium: (1) competition at extracellular sites, probably at the membrane; and (2) intracellular competition at sequestration sites. They confirmed the increased tension of arteries in a potassium-free medium (Hendrick and Casteels, 1974) and showed that magnesium decreases arterial sensitivity to calcium in both high- and low-potassium solutions, but increases the maximum calcium-induced response in high-potassium solution.
Changing magnesium and calcium concentrations affects the vasocontractile responses to hormones. At such low rates of magnesium infusion in dogs as to barely affect arterial resistance, the arterial contraction produced by injected catecholamines was substantially reduced (Haddy, 1960; Frohlich et al., 1962). Suspension of arterial strips in media lacking both calcium and magnesium resulted in almost no contractile response to such agents as acetyl choline, angiotensin, or epinephrine; restoring the calcium but not the magnesium markedly increased the vascoconstriction (Altura and Altura, 1978, 1976/1980) (Fig. 6-5)
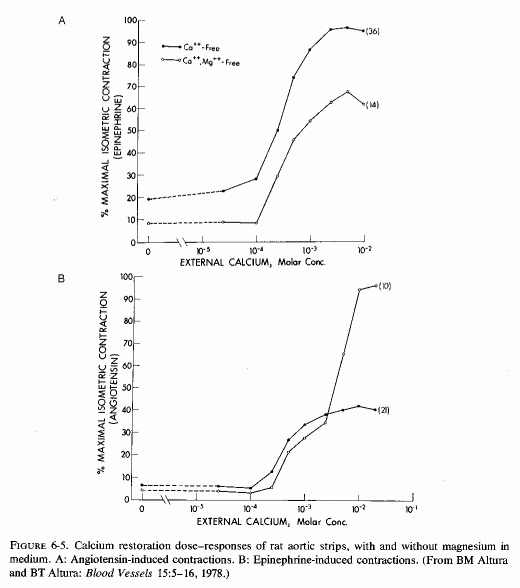{kind=link}
The converse effect, that of the vasodilatory effect of high magnesium concentrations (Haddy, 1960; Haddy and Scott, 1965; Scott et al., 1968; Overbeck et al., 1969), seems to be mediated by displacement by magnesium of calcium bound to the cell surface. This has been shown to inhibit calcium influx and to uncouple excitation from contraction in myocardial cells (Langer et al., 1968; Shine and Douglas, 1974), and is probably also true for vascular muscle. Possibly, in this circumstance, the excess magnesium that displaces calcium from surface binding sites allows for fewer depolarizations and less contractility. Also to be considered is the possibility that high levels of magnesium markedly decrease the hypertensive response to angiotension II, as has been shown in rats (Cession et al., 1963).
In contrast to the above observations, and in conflict with the "logical" explanation of mechanisms by which magnesium deficiency causes vasoconstriction and its excess causes vasodilation, there is both experimental and clinical evidence that magnesium deficiency has caused decreased blood pressure(Fig. 6-6). The demonstration by Cantin (1970, l971/l973a,b) that magnesium-deficient rats develop a continuous increment of the juxtaglomerular index (JGI), and of the width of the zona glomerulosa of the adrenal cortex, explains the aldosteronism of magnesium deficiency reported by Ginn (1968) but not the lack of hypertension in Cantin's deficient rats. He commented on the similarity of the JGI changes produced by magnesium deficiency to those reported after adrenalectomy, sodium deficiency, or renal ischemia (Cantin, 1971/1973a), and considered it plausible that increase in the JGI and the widening of the adrenal cortical zona glomerulosa might reflect response to a shift of fluid from the vascular space (with decreased circulating volume), as occult edema developed (Cantin, 1970, 1971/1973b). He suggested that since the rise of the JGI and the adrenal cortical changes developed early (by the 15th day) in his magnesium-deficient rats, the shifts in sodium and potassium content of serum and urine, and the subsequent marked edema (by the 25th day) might indicate stimulation of the renin-angiotensin-aldosterone system (Cantin, 1970). It was postulated that the JGI and adrenal cortical changes were probably mediated by diminution of the arterial pressure of the renal afferent arterioles, which led to stimulation of the renin-secreting, granular cells of the juxta-glomerular area, with subsequent angiotensin production and stimulation of aldosterone secretion (Cantin, 1970; Cantin and Huet, 1973). In the magnesium-deficient rats of Dagirmanjian and Goldman (1970), the systemic blood pressure was unaffected. They found the blood flow to be diminished by as much as 50% in most organs in the deficient rats that survived 40 days, but that there was splanchnic (gut and liver) vasodilatation that earlier had balanced the visceral vasoconstriction (in terms of systemic blood pressure). Early blood flow changes in these rats included increased flow to the adenylhypophyseal area.
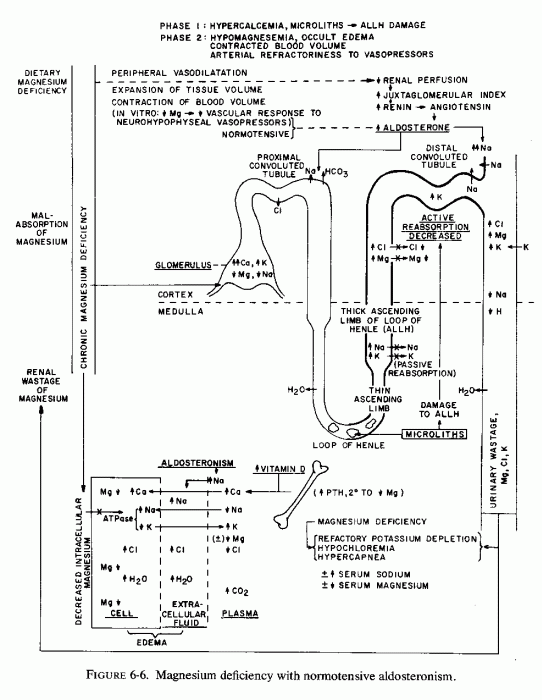{kind=link}
It is suggested (Haddy and Seelig, 1976/1980) that this might be a response to the decreased activity of neurohypophyseal peptides in magnesium deficiency. For example, it has been suggested that magnesium potentiates the contractile response of isolated vascular smooth muscle to vasopressin, oxytocin, and vasotocin, the action of which is magnesium dependent (Somlyo et al., 1966; Somlyo and Somlyo, 1970). Altura (1975a,b; Altura and Altura, 1977) have elucidated the mechanisms by which magnesium enhances the vasoconstrictor response to the vasoactive peptides. Thus, in the absence of optimal magnesium concentrations, the arteries exhibit refractoriness to high levels of the neurohypophyseal peptides. In fact, Altura (1975) clearly showed that the isometric contraction in response to vasopressin was markedly diminished in the absence of magnesium, as compared to its response in the presence of normal magnesium concentration (Fig. 6-7). Responsiveness to angiotensin was absent when both calcium and magnesium were missing from the medium; it was greatly increased when the calcium was restored in the absence of magnesium (Altura and Altura, 1976/1980b). This observation confirms the early in vivo observation that calcium but not magnesium is necessary for angiotension-II-induced hypertension (Cession et al.,1963).
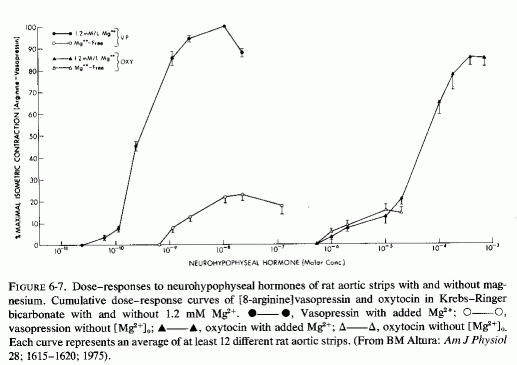{kind=link}
Still another magnesium-vasoactive hormonal interrelationship has been elucidated by Altura et al.(1976, 1976/1980). They have shown that without optimal magnesium in the bath fluid suspending isolated rat arterial muscle, prostaglandin cannot evoke arterial muscle relaxation.
With so many magnesium-related factors influencing arterial contractility, it is not easy to select those that will precipitate either hypo- or hypertension, or produce symptomatic signs of magnesium deficiency without notably affecting the blood pressure. The most dramatic changes in blood pressure mediated by magnesium are (1) the rise that has occurred during iatrogenic hypomagnesemia, produced by replacement of gastrointestinal and renal losses by magnesium-free fluids, which have been lowered by magnesium repletion (Smith et al., 1960; Smith, 1963; Hall and Joffe, 1973); and (2) the hypotension seen with severe hypermagnesemia (Mordes et al., 1975). Less frequently noted is the hypotension of severe magnesium depletion as in children with the recovery syndrome of protein calorie malnutrition (Caddell, 1965, 1967).
In general, gradual or chronic changes in serum magnesium levels are not associated with marked changes in blood pressure. On the other hand, note should be taken of the hypomagnesemic form of aldosteronism (Mader and Iseri, 1955; Mime et al. 1957) that is usually associated with moderately to markedly elevated blood pressure. In contrast, a woman has been reported who had marginal magnesium deficiency, occult edema, signs of latent tetany, and subnormal blood pressure, with intermittent aldosteronism and hyperreninism (Seelig et al. 1975, 1976/ 1979), a syndrome much like that reported by Cantin in rats (Cantin, 1970, 1971/1973).
Whether magnesium deficiency contributes to the hypertension of children with the supravalvular aortic stenosis syndrome that is associated, not only with hypercalcemia, but hyperlipidemia and that has been associated with hyperreactivity to vitamin D (Reviews: Black, 1964; Beuren et al., 1962, 1964, 1966; Seelig. l969b; Seelig and Haddy, 1976/1980) remains to be ascertained. There have been a few instances of hypomagnesemia reported, but the use of milk of magnesia to control the common constipation of this syndrome makes it difficult to interpret the rare reports of magnesium levels. Hypertension and hyperlipidemia have been reported in children and adults with hypervitaminosis D (Frost et al., 1947; Lang and Eiardt, 1957; DeLangen and Donath, 1956; Beuren et al. 1964, 1966; and 24 cases in Appendix Table VIa; low serum magnesium levels have been reported only rarely (Frost et al., 1947; Lowe et al.,. 1954). Dalderup (1960) speculated that the damage of infantile hypercalcemia might be related to cellular magnesium deficiency. Other conditions associated with hypercalcemia and hypomagnesemia in which hypertension is not uncommon include hyperparathyroidism (Pyrah et al., 1966) and hemodialysis with "softened" water (Schulten et al., 1968).
The relationship of the magnesium status to adult hypertensive syndromes is difficult to ascertain. Most of the emphasis has naturally been on sodium/potassium exchanges. The potentiation of the pressor effects of catecholamines by corticosteroids, which was demonstrated by Raab, and correlated with the transmembrane Na/K gradient and blood pressure regulation (Raab, 1959), can also be referred to as regards magnesium/calcium shifts. Both hormones cause magnesium egress from the cells; catecholamines also increase calcium influx.
As has been discussed, hypomagnesemia is common in preeclampsia and eclampsia, and hypotensive as well as anticonvulsive response to magnesium therapy in pharmacologic doses is anticipated. However, still to be proved is whether these responses reflect repair of a deficit or merely vasodilation in response to a pharmacologic agent. Similarly the use of magnesium to control hypertensive crises of renal disease (during the diuretic phase) requires resolution as to mechanism. In hypercalcemic hypertension, renal damage may complicate the diagnostic problem.
As regards essential hypertension, the common use of diuretics that cause renal magnesium loss makes interpretation of serum magnesium levels difficult Holtmeier (1969b) has surveyed cardiovascular and other complications of diuretic treatment of hypertension that might result from magnesium loss and recommends its repletion.
Part II: Chapter 7
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
The foregoing section has dealt predominantly with the evidence that magnesium deficiency can be contributory to arterial lesions (culminating either in sudden death or in chronic atherosclerosis) that are implicated in the cardiovascular diseases of civilization. Raab (1972), in his introduction to a symposium on myocardiology, commented that the current "official" approach to the problem of degenerative heart disease represents adherence to "traditional but outdated concepts that imply a purely, or almost purely coronary vascular origin of fatal myocardial lesions." He referred to evidence that in about half the deaths clinically attributed to "myocardial infarctions," "coronary occlusions," "coronary thrombosis," or "coronary artery disease," no thrombi or vascular occlusions were found on autopsy (Baroldi, 1969, 1970/1972; Spain and Bradess, 1960). He suggested that the term "coronary heart disease" be replaced by one referring to "cardiac hypoxic dysionism," as encompassing the ionic myocardial changes produced in association with the myocardial hypoxia resulting from a decreased oxygen supply (coronary insufficiency) in conjunction with stress-induced hormonal (catecholamine) increased oxygen demand (Raab, 1969). As depicted in Fig. 7-1 (Raab, 1972), hypoxia causes decreased myocardial concentrations of both magnesium and potassium and increased myocardial sodium. This dysionic pattern is contributed to by stress-induced corticosteroid secretion.
{kind=link}
When myocardial levels of magnesium fall, there are many contributory factors. That nutritional imbalances leading to general magnesium deficiency, such as have been described in the introductory chapter on epidemiology, can contribute and can reduce the resistance of the myocardium to stress and to noxious agents seems likely. It might be the extra magnesium that is provided by hard water that is responsible for the much lower incidence of sudden death from ischemic heart disease (IHD) in residents of hard-water areas, as compared with the IHD sudden- death rate in soft-water areas. It is possible that water-magnesium is sufficient to correct (at least partially) the marginal magnesium deficiency that has been shown to be prevalent and increasing in the United States and in Europe. The lower myocardial and coronary magnesium levels found in accident victims in soft-water areas, as compared with those found in comparable subjects in hard-water areas, indicate that an insufficient magnesium intake is reflected by lower myocardial magnesium levels (T. Crawford and M. D. Crawford, 1967; T. Anderson et al., 1973, 1975, 1976/1980). It is important to note that despite the difference in myocardial magnesium levels, plasma magnesium levels in residents of soft- and hard-water areas are the same (T. Anderson et al., 1975, 1976/1980). This is another indication of the unreliability of plasma magnesium as an indication of the total status of magnesium or of its level in vital organs.
Although, on the grounds of logic, one would expect the heart to retain magnesium with avidity-since substantial loss of myocardial magnesium is incompatible with life-cardiac magnesium is actually quite labile. Rogers and Mahan (1959 a,b) reported that in the exchange of plasma magnesium with tissues, there are rapidly and slowly exchangeable forms of magnesium in the tissues of rats. In the heart, liver, and kidney, the exchange was rapid, reaching equilibrium in about three hours. In cows and calves, the equilibration was slower than in rats, but liver, kidney, heart and pancreas similarly showed most rapid exchangeability of 28Mg (Rogers et al., 1964). Page and Polimeni (1972), also working with rat hearts, have demonstrated that about 98% of ventricular cellular magnesium is exchanged at the same but relatively slow rate [disagreeing with Rogers and Mahan (1959 a,b) that the exchangeable portion was "rapidly" exchangeable]. They found that only 2-3% is inexchangeable (Page et al., 1972), in contrast to that of skeletal muscle, 75- 80% of which is exchangeable (Gilbert, 1960). They found that the rate of myocardial magnesium exchange is 0.15 ± 0.02 mM Mg/minute/kg-1 dry ventricle or about 0.21 ± 0.02 pmol/sec/cm-2 of plasma membrane. The rate of exchange is independent of the rate of contraction or the external work done by the ventricle (Polimeni and Page, 1973a,b, 1974). Measurements of the influx and efflux of magnesium and the very low passive permeability of myocardial cells to magnesium suggested that there is probably a carrier-mediated mechanism for its cardiac transport that might be capable of preventing development of unphysiologically high myocardial cellular levels of magnesium (Page and Polimeni, 1972). Most of the 98% of exchangeable myocardial magnesium is presumably present as Mg complexes of the adenine nucleotides: ATP, ADP, and AMP. Less than 15% is associated with the mitochondria or myofibrils (Polimeni and Page, 1973a). The mitochondrial mechanism of magnesium transport has been shown in vitro to cause accumulation of large amounts of magnesium by transporting it across the inner mitochondrial membrane into the matrix (Brierley et al., 1962; Brierley, 1967; 1976). More recent studies show that there are mitochondrial ionophores that mediate magnesium (and other ions) transport (Green et al., 1975) and that such ionophores have been identified in heart mitochondria (Blondin, 1974, 1975). In rat ventricles all of the mitochondrial magnesium is exchangeable with28Mg given intraperitoneally (Page and Polimeni, 1972), and so possibly this may be a means of regulating the amount of ionic magnesium in the cytoplasm. in vivo, myocardial cells accumulate a proportional increase in magnesium in response to stimuli that cause cellular hypertrophy (such as mechanical constriction of the ascending aorta); under such conditions there is also a disproportionate increase in sequestered myofibrillar magnesium (Page et al., 1972). Polimeni and Page (1973b, 1974) comment that a constant cellular magnesium concentration is essential to the myocardial cell. They observe that since a major proportion of cellular magnesium is complexed with the adenine nucleotides, it may be the constancy of the magnesium concentration that is related to the constancy of adenine nucleotide concentrations, which is necessary for normal myocardial cellular metabolism, including ionic exchange and energy production.
The ready exchangeability of almost all myocardial magnesium, and the demonstration of Mg2+-K+, specific mitochondrial ionophores that mediate myocardial mitochondrial magnesium transport, probably explain its rapid uptake, which is demonstrable when it is given as 28Mg Brandt et al.(1958) reported that of all the soft tissues and viscera analyzed from 24 to 48 hours after 28Mg administration (i.v. infusion to rats), the heart took up the greatest proportion of the isotope. [The kidneys, liver, and pancreas took up less, but much more than did the other tissues studied (Fig. 7-2)]. They suggested that finding such marked avidity for the magnesium, in tissues with high enzyme activity, was not surprising, in view of the importance of magnesium in ATP and other enzyme systems (Lehninger, 1950; Green and MacLennan, 1960). The heart's uptake of 28Mg has been shown to be ten times as rapid as that of skeletal muscle (Brandt et al., 1958; Aikawa et al., 1959; Field, 1961; Field and Smith, 1964). In view of their demonstration of the particular avidity of dogs' hearts for 28Mg in dogs, Glaser and Brandt (1959) extended the study to calves and rabbits. The findings were consistent in the three species. They found the greatest avidity for 28Mg in the interventricular septum and in the left ventricle of calves. The authors postulated that the high 28Mg uptake of the septum might reflect the requirement of the conduction system for impulse transmission. The greater septal and left ventricular uptake of 28Mg than in the rest of the heart was reaffirmed in dogs (Glaser and Gibbs, 1962). Lazzara et al. (1963) confirmed the avidity of ventricles and septum for tagged magnesium at 46, 56, and 68 hours after its i.v. administration in dogs. It is of interest that in an analysis of different portions of the heart (of dogs), Burch et al. (1965) found the highest cardiac concentrations of magnesium in the interventricular septum, epicardial myocardium, and ventricles. Rogers et al. (1964) did not specify the portions of the heart scanned, but included that organ as one of those with the highest specific activity 2-6 hours after injection of 28Mg into cows and calves.
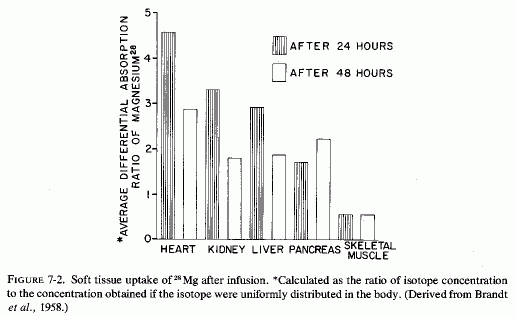{kind=link}
In magnesium-deficient rats, the relative specific activity (the ratio of the specific activity of the tissue to that of plasma) of heart and other metabolically active organs (e.g., kidney, liver, glandular tissue) reached peak levels within 2-3 hours after injection of 28Mg, and remained high through the 22 hours of the study. The values declined after an initial rise in control rats (Field and Smith, 1964). Comparison of magnesium-deficient and control rats given 28Mg before sacrifice at intervals up to 48 hours, showed rapid uptake (at 2-4 hours) that was most marked in liver, heart, and kidney (Chutkow, 1965). All subcellular fractions of the myocardium, from rats kept magnesium deficient for 32 days, exhibited avidity for 28Mg that had been given 12 hours before sacrifice (Ryan et al., 1973). Rats that had been repleted for 18 days before the 28Mg injection did not show greater than control 28Mg uptake, indicating repair of the myocardial deficit within that period of time.
The amount of myocardial magnesium might be the factor that determines cardiac response to the many cardiopathic factors in our environment. Dietary imbalances that increase magnesium requirements at the same time that less is ingested have been shown to lower myocardial magnesium levels. In experimental animals, such short- or long-term magnesium deficiencies have produced arrhythmias, coronary arterial lesions, and light- and electron-microscopic evidence of damage that is intensified by stress. Hormones that stress causes to be secreted (e.g., catecholamines and corticosteroids), and drugs or hormones that cause further loss of magnesium, particularly when associated with retention of calcium (e.g., diuretics, digitalis, vitamin D, dihydrotachysterol), have similar effects.
This brings us to the concept of "pluricausal cardiomyopathy," a term used by Selye (1961, 1969) and Raab (1969, 1972) as preferable to the limiting term "coronary heart disease." They used it to encompass also hormonal and dysionic responses to emotional, as well as drug-induced stresses and metabolic aberrations. Selye (1969) commented that deficiencies in dietary potassium, magnesium, or chloride each predisposes to cardiac necrosis closely resembling that of his electrolyte-steroid cardiac-necrosis (ESCN) experimental model in that all produce extensive, usually multifocal myocardial necrosis. Excessive concentrations of epinephrine like substances in the heart of a young athlete who had died suddenly (Raab, 1943a), and in hearts of patients who had died with angina pectoris and other cardiac dysfunctions (Raab, 1943b), and the similarity of the ECG changes of patients with IHD to those of animals or humans given epinephrine, led Raab to consider stress-induced hormonal (catecholamine and corticosteroid) excess as basic to the disorder he termed cardiac "dysionism" (Raab, 1972). He observed that major shifts in myocardial electrolytes can lead to disturbances in cardiac rhythm, contractility, structure, and ultimately to cell necrosis. His emphasis was on the depletion of intracellular potassium, but he observed that this was usually paralleled by loss of glycogen and magnesium and by entry of sodium into the myocardial cells.
Since experimental magnesium deficiency was first recognized as causing cardiac damage, both functional and morphological, and since development of the electron microscope has permitted demonstration of mitochondrial changes (remarkably similar to those produced by experimental ischemia) that can explain the dysionism referred to by Raab (1969, 1972), the cardiac changes caused by magnesium deficiency are presented before the discussion of the role of magnesium loss in dysrhythmias.
Functional and histologic abnormalities of the heart were demonstrated in magnesium-deficient rodents and ruminants over 40 years ago (Greenberg et al., 1936; Moore et al., 1936) and low cardiac magnesium levels in the failing human heart even earlier (Wilkins and Cullen, 1933). The nature of the damage that is caused by experimental magnesium deficiency, and the protective effects of magnesium administration, have been demonstrated in many animal models of cardiovascular disease (Reviews: Selye, l958g; Bajusz, 1965; Heggtveit, 1965c, Raab, 1969; Lehr, 1969; Rigo, 1971; Rotman, 1971; Szelenyi, 1971; Seelig, 1972; Seelig and Heggtveit, 1974; Seelig and Haddy, 1976/1980). As indicated, the "pure" magnesium deficient heart has histological myocardial lesions that are predominantly perivascular (around the damaged small coronary arteries) and thus probably reflect hypoxia secondary to the early arterial damage.
Light microscopic lesions (including focal myocardial necrosis, exudative inflammation, and varying degrees of calcification and collagen deposition) were seen in rats that were magnesium depleted for 14-36 days, the degree of damage being directly related to the duration of the depletion (Heggtveit et al., 1964; Heggtveit, 1965b,c). A group that was also cold stressed (swimming in ice-water bath for four minutes) twice daily the last two days before sacrifice exhibited the most severe damage; they were the only rats to exhibit grossly evident cardiac damage. Many of the myocardial lesions were perivascular, surrounding small ramifications of the coronary arteries, but this was not a consistent finding. Primary arterial damage, other than edema of the endothelium, was not noted. Ultrastructural changes in the myocardium were most pronounced in and around the areas of necrosis. Like the magnesium-deficient rats reported by Nakamura et al. (1961) that had swollen mitochondria after 12 days of magnesium deficiency, those of Heggtveit et al. (1964) also showed mitochondrial or sarcosomal swelling and distortion (at 14 days). There was vacuolization of enlarged sarcosomes, clumping of cristae, and progressive deposition of electron-dense material, which eventually filled the entire saracosome or mitochondrion especially in the magnesium-deficient stressed rats (Fig. 7-3). Rats given the same diet, but with magnesium supplements, developed no cardiac lesions, whether or not they were cold stressed.
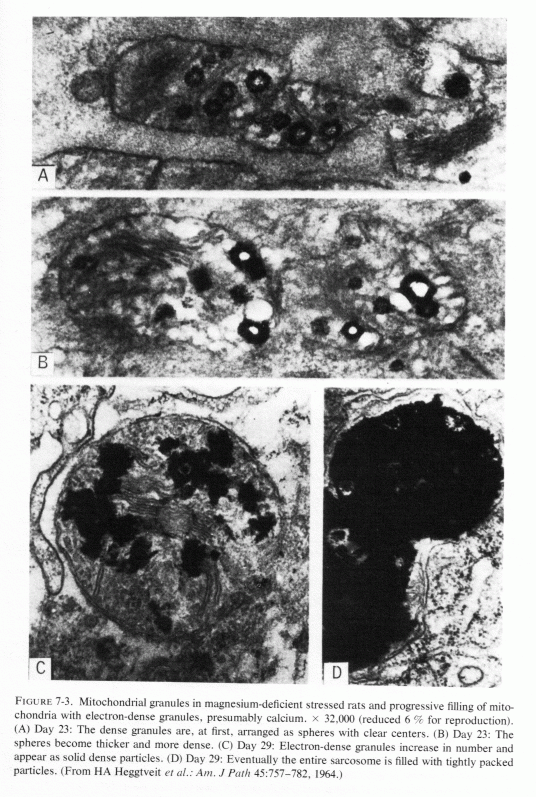{kind=link}
Fragmentation and loss of myofilaments (which make up the myofibrils) both accompany and follow the sarcosomal changes. Thus, there is disruption of "Z" bands and "M" lines, with spaces within the myofibers. Aggregating within these spaces (corresponding to vacuoles seen by tight microscopy) are dilated components of the sarcoplasmic reticulum, damaged sarcosomes and ground substance, lipid droplets and glycogen particles. Finally, the sarcolemmal membrane ruptures or disappears, and the altered sarcoplasmic constituents spill into the interstitial space, where they are ingested by macrophages aligned alongside necrotic muscle cells (Heggtveit, 1965c).
Mishra (1960b), who had found that the mitochondrial fraction of hearts from magnesium-deficient rats was diminished, reasoned that such a loss, which is linked to oxidative phosphorylation, might be responsible for defective ability of magnesium-deficient mitochondria to maintain ionic gradients and for metabolic and respiratory cell injury leading to myocardial necrosis. DiGiorgio et al. (1962) proposed that since the amount of magnesium in the distorted cardiac sarcosomes was the same (or even more) in the magnesium-deficient than in the control rats, possibly it was in a form unsuitable for coupling of oxidation to phosphorylation.
Ultramicroscopy has shown that magnesium deficiency for as little as 12-14 days has caused cardiac mitochondria to swell (Nakamura et al., 1961; Heggtveit et al., 1964; Heggtveit, 1965b,c); that such swelling is not physiologic, such as occurs during ionic flux (Fig. 7-4A), but is pathologic (Fig. 7-4B). It is associated with mitochondrial disruption and disorganization. The electron dense particles probably consist of calcium (e.g., as phosphate crystals). Possibly some of the mitochondrial magnesium is similarly made unavailable (Jennings, 1969; Seelig, 1972). Such redistribution of the calcium and magnesium ions, taking them and the inorganic phosphate out of the pool available for oxidative phosphorylation, might be contributory to irreversible mitochondrial damage. It must be noted that the mitochondria from a magnesium-deficient rat that had marked mitochondrial and sarcosomal calcium granular deposition (Heggtveit et al.., 1964; Heggtveit, 1965b,c) were from a rat that was cold stressed.
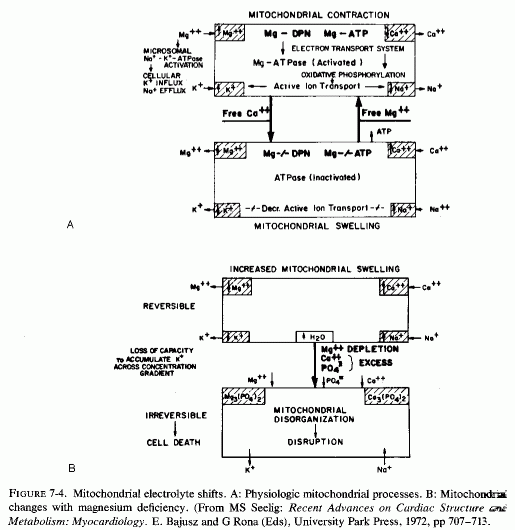{kind=link}
Heggtveit (1965c) has reviewed the data correlating the close interdependence between mitochondrial structure and function and has observed that early sarcosomal alterations are fundamental to the evolution of the cardiac necrosis of magnesium deficiency (Heggtveit et al., 1964). He noted that the calcium accumulation occurring in magnesium deficiency begins before the cell dies. A recent in vitrostudy provides evidence that magnesium modulates calcium uptake in cardiac mitochondria (Silver and Sordahl, 1976/1980). Respiration-supported calcium uptake by rabbit-heart mitochondria in magnesium-free medium was almost double that in the presence of magnesium. Furthermore, in the absence of magnesium, the calcium crystals in the mitochondrial matrix were needlelike. On addition of magnesium to previously magnesium-free suspensions, they underwent transformation into an apparently destructive granular type with dendritic crystals that obliterated the internal mitochondrial structure (Fig. 7-5). In the presence of magnesium, spheroidal-amorphous calcium crystals form in the mitochondria. Silver and Sordahl (1976/1980) suggest that the magnesium modulation of the calcium uptake, and its influence on the shape of the crystals, is consistent with the protection afforded by magnesium against the necrotizing effects of calcium on myocardial cells when magnesium levels are low (Janke et al., 1975; Lehr et al., 1975).
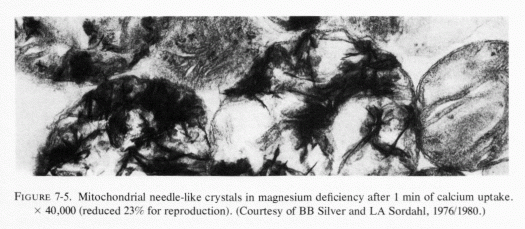{kind=link}
There is loss of myocardial magnesium from hearts of experimental animals with coronary ligations after asphyxia (Table 7-1) and from infarcted areas of human hearts (Table 7-2). Why the drop in myocardial magnesium was greater after transient ischemia (following reestablishment of circulation) than it was in dog hearts with permanent ischemia (Jennings and Shen, 1970/1972) was not explained, but it is in accord with the short-term (up to 10 minutes) findings of Hochrein et al. (1967) with asphyxiated guinea pigs and with the long-term study of electrolyte changes in infarcted versus noninfarcted areas of the heart from 5 hours at intervals to 30 days (Table 7-3, Matyushin and Samartseva, 1972). It is conceivable that the short-term fall in magnesium, reaching almost normal levels 2½ minutes after cardiac arrest, might reflect formation of unavailable (possibly phosphate precipitate) magnesium within the mitochondria (Seelig, 1972).
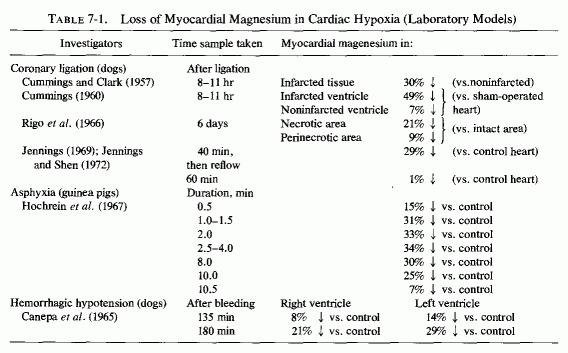{kind=link}
{kind=link}
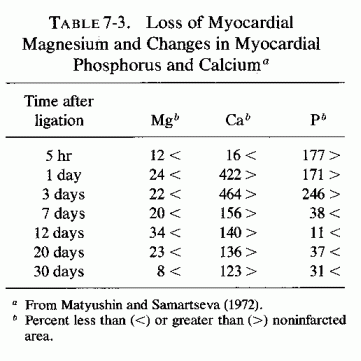{kind=link}
Jennings et al. (1965,1969) have shown that myocardial cells show some mitochondrial damage (some loss of cristae and matrices), some loss of glycogen and some margination of chromatin material within 15 minutes after coronary occlusion. Restoration of blood flow by no more than 18 minutes allowed for resumption of normal structure and function (back to aerobic from anaerobic metabolism). Longer periods of ischemia resulted in irreversible mitochondrial damage, with loss of cristae, disruption of limiting membranes, and intramitochondrial granules (after 40 minutes of ischemia). The dead or dying cells exhibit loss of magnesium, potassium, and acid-soluble phosphate, and gain of sodium, chloride, and water, an electrolyte distribution similar to that of extracellular fluid (Jennings et al., 1969, 1970). Later, the calcium and phosphorus levels rise (Jennings and Shen, 1970/1972; Shen and Jennings, 1972), probably as calcium phosphate granules form. Jennings (1969) has suggested that crystallization or binding of essential co-factors such as phosphate, calcium, and possibly magnesium in the granules might contribute to irreversible mitochondrial failure. A. Schwartz (1971/1972) commented that mitochondria have the ability to sequester large amounts of calcium, and that if enough calcium interacts with the mitochondrial membranes, there is significant uncoupling of oxidative phosphorylation. Shen and Jennings (1972) demonstrated that ischemic injury causes abnormal calcium uptake as dense intramitochondrial granules, which are an important feature of irreversible cellular injury.
The later, lesser rises in myocardial magnesium that occur in hypoxic hearts probably must be otherwise explained than by accumulation of magnesium phosphate crystals or granules in the mitochondria. Page et al. (1972) have shown that myofibrillar magnesium and mass increases, and the ratio of mitochondrial volume to cell volume decreases in rabbit hearts with mechanical interference with left ventricular outflow, If such myofibrillar sequestration of magnesium occurs in the surviving cells in the area in which ischemia has been induced, perhaps this explains at least partially the later rise in myocardial magnesium levels.
Heggtveit (1965c, 1969) and Heggtveit and Nadkarni (1971), in their reviews of electron microscopic findings of myocardial ischemia, considered the similarities in mitochondrial changes to those of magnesium depletion and catecholamine cardiopathy, which Lehr and his associates had correlated with early loss of myocardial magnesium and accumulation of calcium (Lehr et al., 1966; Lehr, 1969). Heggtveit (1969) pointed out, however, that early nuclear changes are characteristic of ischemic injury, whereas nuclear chromatin clumping occurs only late, after severe sarcoplasmic damage of magnesium deficiency. He commented that correlation of ultrastructural data with biochemical findings confirms the importance of catecholamine release and ionic shifts (early loss of magnesium, potassium, and phosphate with influx of calcium, sodium, and water) in the early evolution of ischemic myocardial damage. Poche (1969) reported that capillary endothelial swelling, with reduction in luminal caliber of the microcirculation, is significant in the pathogenesis of multifocal hypoxic myocardial necrosis. Such endothelial swelling has been reported in magnesium deficiency (Heggtveit 1965c; Hungerford and Bernick, 1976/1980), as have endothelial and medial proliferation. These arterial changes of "pure" magnesium deficiency, thus, might contribute to the hypoxia-like myocardial lesions seen in magnesium deficiency, and might contribute to the decreased resistance of the myocardium to stress factors, such as Heggtveit (1969) suggested might "condition" a chronically ischemic heart to severe response to subsequent acute episodes.
Raab (1943b, 1966, 1969) was the first to point out that catecholamines increase cardiac work and oxygen consumption to the extent that relative hypoxia is produced, particularly in the presence of coronary disease that prevents adequate oxygenation. Relative cardiac hypoxia is also produced with cardiac overload (Hochrein and Lossnitzer, 1969) with similar consequences: stress-induced dysionic status in the myocardium, which leads to functional and finally structural abnormalities in the heart that can result in sudden death from arrhythmias, cardiomyopathies that can lead to chronic heart disease, or the more widely recognized "coronary heart disease." Although he placed major emphasis on the loss of potassium from myocardial cells, he observed that rats stressed by isolation also had low myocardial magnesium levels (Raab et al., 1968), and that patients who had died with ischemic heart disease also had low myocardial magnesium levels (Raab, 1969). He considered the catecholamine release a major mediating cardiopathic response to stress, but called attention to evidence that catecholamine mobilization of free fatty acids from adipose tissues is dependent on the presence of glucocorticoids (Maickel et al., 1966). Thus, he considered the stress release of catecholamines and corticosteroids additive in cardiopathic potential.
It is thus particularly unfortunate that acute coronary occlusion is a very stressful event that stimulates secretion of both of the major groups of adrenal hormones, those of the cortex and the medulla. As regards the catecholamines, such secretion takes place, not only from the adrenal medulla, but also within the heart itself, which synthesizes, stores, and releases norepinephrine (Raab and Gigee, 1955; Braunwald et al., 1964).
Much work has been done on the nature of the gross, histological, and electron- microscopic myocardial necrosis produced by high doses of the potent β-adrenergic amine, isoproterenol, since the work of Rona et al. (1959), which was shown the same year to be intensified by mineralocorticoids (Chappel et al., 1959). Ferrans et al. (1964, 1969), using the high dose (85 mg/kg) that consistently produces large infarction-type lesions, found that mitochondrial swelling, vesiculation, and crystolysis developed early, and myofibrillar degeneration later. Zbinden and Bagdon (1963) found that even with relatively low doses, the myocardial lesions occurred regularly and were located predominantly at the interventricular septum, the apex, and the wall of the left ventricle. The location of the lesions at the sites that had been shown to have the greatest affinity for28Mg (Glaser and Brandt, 1959; Glaser and Gibbs, 1962) and to have the highest magnesium concentration (Lazzara et al., 1963; Burch et al., 1965), and the similarity of the ultramicroscopic lesions to those produced by magnesium depletion are inferential evidence that the catecholamine-induced myocardial might be mediated by loss of myocardial magnesium. Lehr et al. (1966), using small enough doses of isoproterenol (5.25 mg/kg) to produce disseminated myocardial necrosis, rather than grossly evident necrosis, proved the first myocardial changes to be loss of magnesium and phosphorus increased calcium; sodium and potassium changes occurred later (Table 7-4). The decrease in magnesium in the myocardium was demonstrable as early as one hour after isoproterenol injection, even preceding the mitochondrial changes that were evident at two hours. In view of the importance of magnesium in oxidative phosphorylation, it is not surprising that similarly small doses of the catecholamines caused its depression in cardiac mitochondria (B. Sobel et al., 1966).
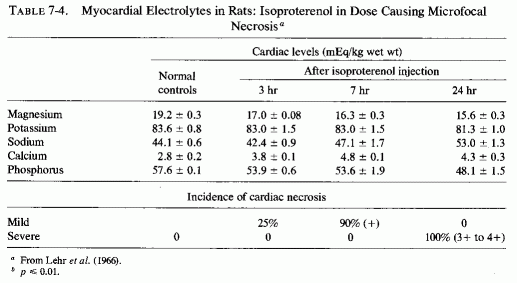{kind=link}
There is another magnesium/catecholamine interrelationship that should be considered. Magnesium and calcium have reciprocal effects on storage or release of catecholamines from adrenergic granules in the adrenal medulla. Mg-ATP stimulates amine incorporation in adrenal medullary granules (Carlsson et al., 1963). Calcium stimulates and magnesium inhibits release of catecholamines from the granules. (W. Douglas and Rubin, 1964; J. Burn and Gibbons, 1964). Since catecholamine granules, epinephrine, or related substances have been demonstrated in the myocardium (Raab, 1943a,b; Potter and Axelrod, 1963b), particularly in hearts from patients with angina pectoris (Raab, 1943b), the observation that in vitro addition of magnesium stabilizes the catecholamines in the heart, preventing releasing of norepinephrine, (Potter and Axe 1963a) might be significant in the clinical situation.
In his thesis on the effects of magnesium deficiency in the rat, C. Johnson (1965) showed that the adrenal medullary levels of epinephrine fell: 23% decrease after 8 days of deficiency and 46% decrease after 12 days of deficiency. Possibly this reflects increased release of epinephrine from the adrenal medullary granules in magnesium deficiency. The same rats also exhibited a slight increase in myocardial catecholamine levels that was associated with low cardiac magnesium and ATP levels.
In addition to the increased output of catecholamines in response to stress, the secretion of corticosteroid hormones is also increased. Selye approached the problem of cardiovascular disease associated with stress from the standpoint that mineralocorticoid secretion was predominantly to blame, particularly in subjects with dietary excesses of sodium, phosphate, and sulfate (Review: Selye, 1958f). In the historical introduction to his 1958 monograph, Selye referred to the early work on the importance of ionic interactions for the function of cardiac muscle in vitro, which led to the recognition of the advantages of physiologically balanced perfusion electrolyte solutions over saline. He reviewed the discovery that had been made at the turn of the century of "acute interstitial myocarditis" for which no cause was identifiable, and commented on the similarity of those lesions to those discovered about the same time (1904) to be produced by experimental overdosage with cardiac glycosides. When irradiated ergosterol preparations became available in 1929, he found that intoxication with sterols of the vitamin D group also produces generalized arterial calcification and focal myocardial necrosis and calcification (Selye, 1929). He noted that all of these myocardial disorders, including that caused by potassium deficiency (Schrader et al., 1937) were characterized by focal necrosis and by inflammatory infiltration (similar to that reported by others using magnesium-deficient diets, supra vide). Then he found that multiple doses of the mineralocorticoid desoxycorticosterone (DOC) caused minute myocardial necrosis in rats, an effect attributed to loss of potassium (Darrow and Miller, 1942). Chicks, fed a ration that was rich in sodium chloride, were more susceptible to DOC-cardiotoxicity (and nephrotoxicity) (Selye and Stone, 1943), and sodium chloride aggravated myocardial necrosis caused by potassium deficiency (Cannon et al., 1953). Selye (1958f) noted that in the "control" potassium-deficient rats in the latter studies, the chloride salt of magnesium had been used as a "filler," in place of the sodium salt, magnesium's protective role not then being generally known. Development of the electrolyte (sodium phosphate)-steroid (mineralocorticoid)-cardiac necrosis (ESCN) model permitted demonstration of some of the factors that intensified or mimicked the myocardial, noninfarctoid lesions. It also permitted investigation of factors with cardioprotective properties (Selye, 1958f). Sodium salts of phosphate and sulfate intensified the lesions; magnesium and potassium chlorides were protective (Selye, 1958 a,d,g, 1961, 1969, 1970b; Selye and Mishra, 1958; Selye and Gabbiani, 1965). Because the ESCN-like lesions could be produced by unrelated agents-cardiac glycosides, vitamin D derivatives, epinephrine, stress-as well as by deficiencies of magnesium or potassium or both, Selye postulated that there must be a common pathway. Also noted was the efficacy of chloride salts of magnesium and potassium against many cardionecrotizing agents (Bajusz and Selye, 1960a). In his surveys of the evolution of the concept that stress contributes to cardiovascular diseases, Selye (1961, 1970a,b) described experiments that, depending on conditioning factors, including stress and the "stress hormones" (ACTH. corticosteroids, and the catecholamines), can produce or prevent cardiovascular lesions. He investigated the importance of mineralocorticoids in the pathogenesis of hypertension, edema, and myocardial lesions (in animals and in human disease) and showed that glucocorticoids that lack a significant mineralocorticoid component do not intensify the ESCN. It is noteworthy that chronic hypercorticism that is associated with sodium and water retention is associated with renal loss of both potassium and magnesium (Review: Massry and Coburn, 1973), and thus functions to increase levels of the "conditioning" cation, sodium, while causing loss of the "protective" cations, potassium and magnesium.
That such a combination of responses can have serious consequences is indicated by the extraordinary potentiation of acute isoproterenol-cardiotoxicity by pretreatment with DOCA and saline (Guideri et al., 1971, 1974, 1978). Such conditioning of the rats resulted in death from fibrillation within 15 to 30 minutes after 150 µg/kg to 0.1 mg/kg of isoproterenol subcutaneously, an amount far below the minimally toxic dose (5 mg/kg) used to produce microfocal necrosis in earlier studies of isoproterenol alone (Lehr et al., 1966; Lehr, 1969) or in combination with mineralocorticoids (Chappel et al., 1959). The myocardial electrolyte pattern showed significant accumulation of sodium and loss of potassium, magnesium, and phosphate. As for the glucocorticoids, the secretion of which is also increased in stress, they further contribute to the metabolic abnormalities by the dependence of catecholamine metabolization of free fatty acids on their presence (Maickel et al., 1966).
Catecholamine levels were not measured in the magnesium-deficient rats (dietary magnesium: 12 mg/g diet for seven weeks) that had much greater degrees of cardiac necrosis when cold stressed than did control cold-stressed rats fed a standard magnesium-supplemented deficient diet (Mishra, 1960e; Heroux et al., 1971/1973). In the latter series (Heroux et al.), four of the eight stressed rats also had 1+ to 2+ cardiac damage (compared to 1+ to 3+ cardiac lesions in all seven of the magnesium-deficient rats). The investigators considered the possibility that the control diet might have been suboptimal in magnesium, and that the control rats might resemble humans on suboptimal magnesium intakes in their susceptibility to myocardial damage of stress.
An interesting model of genetic cardiomyopathy, developed in dystrophic hamsters, might help in elucidating some of the myocardial interrelationships with magnesium and catecholamines. These hamsters, that consistently develop focal myocardial degeneration and myolysis between the 30th and 40th days of life, exhibit decreased magnesium and increased calcium in their myocardium even before the lesions develop (Bajusz and Lossnitzer, 1968), and increased levels of cardiac norepinephrine not long thereafter (Angelakos, 1968; Table 7-5). The low magnesium levels did not persist, but the calcium levels rose markedly in the 56- to 71-day-old cardiomyopathic hamsters, at a time when the norepinephrine levels had risen further (Angelakos, 1968; Angelakos et al., 1970-1972). By the time heart failure had ensued (120 days), the norepinephrine levels had dropped to half the control (young animal) levels, but to a quarter that of the same-age controls (Angelakos, 1968). Cardiac catecholamine stores also decrease once heart failure develops in other experimental models and in human heart disease (Angelakos et al., 1969).
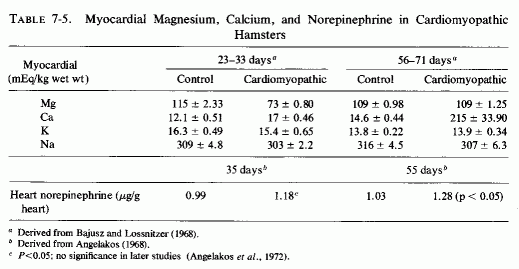{kind=link}
That catecholamines exert a lipolytic effect and increase circulating free fatty acids has been considered earlier. The significance of this on the response of the heart can be considerable. Balazs et al. (1962) and Balazs (1972) have shown that injected catecholamines or stress-induced catecholamines secretion is much more likely to cause serious myocardial damage in obese than in normal rats. This supports the contention that catecholamine-induced lipolysis can be a significant risk factor, especially in overweight patients.
Although free fatty acids can be utilized by the myocardium as an oxidative substrate, there is growing evidence that high levels of free fatty acids (e.g., mobilized by catecholamines) are cardiotoxic (Rosenblum et al. 1965; Opie, 1969; Hoak et al., 1970-1972) and can interfere with myocardial function, especially in association with hypoxia (A. Henderson and Sonnenblick, 1970, 1970/1972; Shug and Shrago, 1973). Opie (1969) has evaluated the relative importance of glycolytic and fatty acid metabolism of the heart, and points out that with excesses of fatty acids and triglycerides, there is substantially increased cardiac oxygen consumption. This can intensify the relative myocardial hypoxia caused by stress, especially in the presence of coronary insufficiency.
The possibility that high levels of free fatty acids in the blood might contribute to symptoms of alcoholics by binding magnesium has been mentioned earlier, as has the favorable response to magnesium of hyperlipemic patients with occlusive arterial disease (Seelig and Heggtveit, 1974). It is possible that postinfarction arrhythmia might be related either to excessive catecholamine release in response to the stress of the cardiac injury, or to catecholamine-induced increase in circulating fatty acids. The catecholamines are apt to lower the cardiac magnesium levels; the free fatty acids might bind magnesium in the blood. Perhaps increased myocardial lipids, such as have been attributed to catecholamine-lipid mobilization in rats injected with sympathomimetic agent (Ferrans et al., 1964; 1969) and in the ESCN model, (Prioreschi, 1966), might be the result of inactivation by the intramyocardial fats of cellular magnesium. Direct evidence that a variety of dietary fats (corn oil, peanut oil, olive oil, pork fat, butter, and chicken fats, as well as saturated and unsaturated fatty acids) greatly increase the sensitivity of the rats to ESCN has been provided (Selye, 1961; Selye et al., 1969).
The cardiovasopathic diet developed by Sos and his co-workers that produces spontaneous myocardial infarctions includes saturated fats and hyperlipemic and hypercalcemic nutrients. Although the diet produces only minor serum electrolyte changes it substantially lowers myocardial magnesium levels. A similar diet, which was described as thrombogenic and which resembles the diet developed by Vitale and his co-workers to produce atherogenesis, incorporates propylthiouracil and cottonseed oil; when Na2HPO4 is added it causes nonocclusive infarctoid myocardial lesions (Savoie, 1972a,b, 1975). Anticholesterolemic agents lower the blood lipids, but are ineffective in protecting against the myocardial necrosis (Savoie, 1972b). The potassium-sparing agents (triamterine and spironolactone), and, to a lesser degree, potassium chloride, are partially protective against the cardiac lesions but not against the hyperlipidemia; only magnesium chloride prevented the cardiac necrosis (Savoie, 1972b). Similarly, amiloride, another potassium-sparing agent, inhibits the development of cardiac lesions produced when corn oil is added to the diets of rats on the ESCN regimen, although the protection is not complete (Kovacs et al., 1969; Solymoss et al., 1969). Ultramicroscopy showed that there was still evidence of myofibrillar damage and mitochondrial lipid droplets, although marked focal lipid accumulations were prevented (Kovacs et al., 1969). Also, the blood lipids were not normalized (Solymoss et al., 1969). Savoie (1971b) demonstrated protection against myocardial necrosis in a comparable model also for triamterene and spironolactone, the agent that blocks aldosterone under conditions of chronic hypersecretion (Massry and Coburn, 1973), such as is seen in heart disease (H. Wolff et al., 1957) and in primary aldosteronism in which it has been associated with magnesium loss (Mader and Iseri, 1955; Milne et al., 1957). All of these potassium-sparing agents also protect against the hyperlipidemic cardiac necrosis that is intensified by stress, epinephrine, or digitalis (Savoie, 1971a,b). Although amiloride did not prevent the lowering of myocardial magnesium in the ESCN+ corn-oil model that causes severe damage in a few days, and in fact actually lowered it somewhat (Solymoss et al., 1970) the potassium-sparers (amiloride and triamterene) also exert some magnesium-sparing activity (Hänze and Seybirth, 1967; Heidland et al., 1970, 1973; Walker et al., 1972). Whether this effect contributed to their partial efficacy in the hyperlipidemic +Na2HPO4 model, in which they were more effective than potassium chloride, but less effective than magnesium chloride (Savoie 1972b) should be further studied. It is of interest that in this less acute model, which possibly is more similar to the situation produced by human dietary indiscretions, the addition of sodium phosphate to the hyperlipidemic diet caused substantial lowering of both myocardial and serum magnesium levels by day 21 (Fig. 7-6, Savoie, 1975). (Note should be taken that in the hyperlipidemic and in the high phosphate-fed rats, the serum magnesium level did not reflect the drop in the myocardial levels.) Savoie (1975) considers the critical factor in the Na2HPO4 and hyperlipidemic rats to be their susceptibility to mitochondrial dysfunction caused by the low myocardial magnesium levels. Since the Na2HPO4 and the hyperlipidemia alone also lowered the myocardial magnesium levels markedly, it seems plausible that each imbalance could increase the susceptibility of the heart to stresses or other agents that further lower myocardial magnesium levels, with resultant arrhythmias or necrosis. This investigator has recently correlated the protective effect of magnesium in this model with its lowering of free cholesterol levels in the heart (Savoie and DeLorme, 1975/1980).
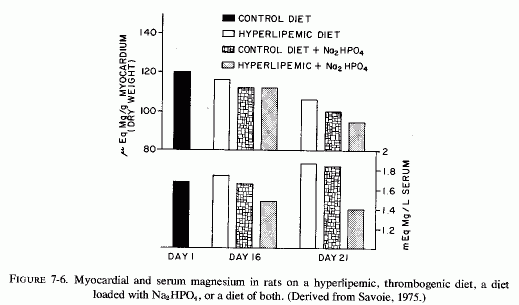{kind=link}
Severe multifocal myocardial and renal necrosis, produced in parathyroidectomized rats given Na2HPO4 is preceded by markedly lowered myocardial (and renal) magnesium levels (Lehr et al., 1966; Lehr, 1969). In addition, the microcirculation of the heart is damaged; many arterioles and precapillaries show loss of the normal architecture, with the presence of arterial and periarterial PAS-positive material. After a single Na2HPO4 load, there is clumping of myocardial sarcosomes, edema, and disturbances in the normal myofibrillar pattern. After two sodium phosphate loads (at 24 hours), the mitochondria are swollen and exhibit disappearance of cristae and formation of granular debris; there is also margination of nuclear chromatin. Lehr (1969) commented that the consistent, significant early shifts in tissue cations (decreased magnesium and phosphorus, and increased sodium and calcium) in this experimental model, as well as in other very different models (e.g., catecholamines, ESCN, and cardiac overload) and the fact that the ionic shifts precede morphological damage, suggest that they are the cause, not the consequence of the myocardial damage.
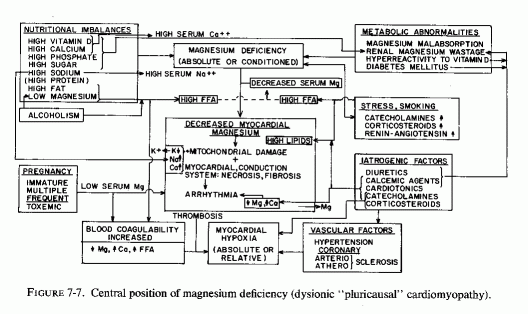{kind=link}
As indicated in the sections on the effects of magnesium deficiency on the arteries, early damage to the small coronaries with narrowing of their lumina is characteristic of magnesium deficiency. Such myocardial arterial disease is not what is referred to by "coronary disease," but it certainly contributes to microfocal areas of hypoxia, which can give rise to the microfocal necroses, infiltration, and fibrosis that have been described in magnesium-deficient animals (Review: Seelig and Haddy, 1976/1980). It is provocative that Lehr (1965b, 1969, 1972) and his co workers (Lehr et al., 1966, 1970/1972, 1976/1980), who proposed that the loss of myocardial magnesium might contribute to the disseminated myocardial necrosis caused by dissimilar agents (including catecholamines and sodium phosphate loading of corticosteroid-treated or parathyroidectomized rats), had also implicated damage to the microcirculation (Lehr, 1965a, 1966, 1969). If magnesium nutritional deficiency or drug-induced myocardial loss is a basic contributory factor [and it has been shown to predispose also to the dysionism (decreased potassium and increased sodium), as well as to increased accretions of mitochondrial calcium (Lehr, 1969, 1972; Review: Seelig, 1972)], then Lehr was correct in both postulates.
The loss of magnesium from the myocardium occurs so soon after hormonal or other challenge as to be suggested as a chemical means of detecting one of the earliest characteristics of myocardial damage (Lehr et al. 1976/1980). Its early loss after experimentally induced hypoxia (Table 7-6, from Hochrein et al., 1967) and from the heart after ischemia from coronary ligation suggests that the loss of magnesium from the fine structures of the myocardium are probably basic to the myocardial damage. Nutritional magnesium deficiency can result in early mitochondrial loss and damage directly or secondarily, as a result of focal ischemia from the narrowing of the intramyocardial vessels. Whether the hypoxia is relative or absolute, there are both increased net loss (efflux) of magnesium from myocardial cells and decreased magnesium influx during hypoxia (Polimeni and Page, 1974).
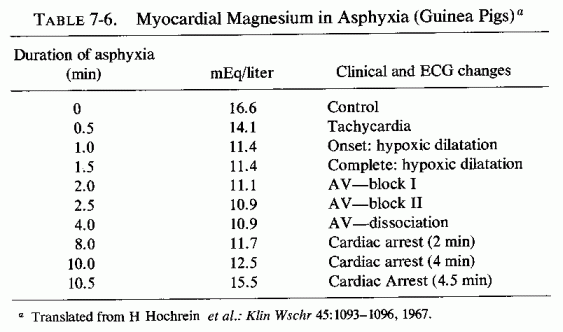{kind=link}
Evidence is presented that common to a wide variety of experimental models of experimental cardiomyopathy is early loss of myocardial magnesium. In his discussion of the lesions produced not only by isoproterenol, but by low doses of other catecholamines, and in other cardiopathic models [i.e., parathyroidectomy and phosphate loads, mineralocorticoid and phosphate loads (ESCN model of Selye), and hypoxic heart failure (Hochrein and Lossnitzer, 1969)], Lehr (1969) proposed that the common denominator was magnesium loss from the myocardium. He proposed that the magnesium loss is primary, and deserves closer scrutiny in view of its importance in the vital energy processes of the cell. He further proposed that its depletion might contribute to the initiation of cellular injury.
It is the premise of this section that the underlying factor-that which determines whether the individual will withstand stress or other potentially cardiopathic factors-is the adequacy of magnesium in his heart. Some might develop dysrhythmias (possibly suddenly fatal) as a result of inadequacy of the damaged magnesium-dependent mitochondrial enzyme system to maintain normal ionic equilibrium, or as the conduction system is affected or myocardial cell excitability is increased by a low magnesium/calcium ratio (infra vide). Others might develop coronary arterial disease, including hypertension, and microscopic or gross myocardial lesions that lead to chronic heart disease. The foregoing section on the lability of cardiac magnesium shows that magnesium can be readily lost from the heart, but that it can also be quickly repleted. Barnes (1962) showed that puppies kept on magnesium deficient diets for two months lost proportionally as much magnesium from the heart as they did from bone, the major magnesium store of the body. The lower myocardial magnesium levels in residents of soft-water areas than in dwellers in hard-water areas (see Chapter 1) supports the contention that long-term suboptimal magnesium intakes are associated with loss of magnesium from the heart, and with a high incidence of sudden death from IHD.
Part II: Chapter 8
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
There are several clinical conditions associated with cardiac abnormalities that resemble those produced by experimental magnesium deficiency or that cause loss of myocardial magnesium. Bajusz (1965b) refers to experimental necrotizing cardiomyopathies to describe a variety of degenerative processes that are more or less confined to the myocardium. He commented that the disease is characterized by subendocardial necrotizing foci, usually without significant disease of the coronary vessels, although comparable disorders can be produced by thrombogenic diets. He observed that "coronary heart diseases" should be classified as primary or secondary cardiomyopathies that result from vascular factors (e.g., coronary artery spasms, local microcirculatory changes), factors that directly affect myocardial metabolism and the susceptibility or resistance of the myocardium at the time of potentially cardiotoxic episodes. He pointed out that, in addition to stress situations, hormones, and age, cardiomyopathy-conditioning factors include sodium excess and deficiencies of chloride, and especially of potassium and magnesium. Bajusz (1969) has stated that the loss of these two cations from the myocardial cells (that are associated with early ultrastructural changes such as enlargement and vacuolization of the sarcoplasmic reticulum and mitochondrial degeneration) seem to be important components of many, if not all types of disturbances in cardiac metabolism, resulting in myocardial degeneration, heart failure, or fatal conduction defects.
Cardiomyopathies (and dysrhythmias not caused by diagnosed ischemic heart disease) are generally classified by clinical manifestations or by pathologic characteristics. In a criticism of any classification of primary (or secondary) cardiomyopathies that is dependent on postmortem diagnosis, Mattingly (1970) suggested that what is needed is greater effort directed toward recognition of early clinical features, search for etiologic factors, study of biochemical as well as hemodynamic alterations, and search for iatrogenic factors in the pathogenesis of and for control of this disease. Because magnesium deficiency or loss can be correlated with many of the cardiomyopathies for which a cause has been established, perhaps it participates in those considered primary or idiopathic.
The term "cardiomyopathy" was introduced by Brigden (1957) to indicate isolated noncoronary myocardial disease, present without significant disease of other systems of the body. He included instances of familial disease, amyloidosis, alcoholism, a few postpartum, and several that seemed to be due to infection. Restriction of the term to isolated myocardial disease might be unwise, since it excludes very similar cardiomyopathies associated with diseases that might provide clues to a common pathogenic factor. In subsequent surveys, the term has been used as synonymous with "myocardial disease," to indicate dysfunction of the muscular pump that is not the result of structural deformity of the heart, hypertension, or coronary atherosclerosis (Editorial, Brit Med J, 1969). Goodwin (1970) classifies them as congestive and hypertrophic. Hudson (1970) suggests four features that characterize cardiomyopathies: cardiomegaly, endocardial thickening, mural endocardial thrombus, and myocardial scars. He considers the fibrotic cardiomyopathies (sometimes accompanied by foci of myocardial calcification) to be likely to be related to hyperreactivity to vitamin D in infancy. He cites myocardial fibrosis in adults as the commonest form of idiopathic cardiomyopathy; it can be familial or occur as isolated cases. Hypertrophic obstructive cardiomyopathy is also familial in some instances, and Hudson (1970) considers it congenital. It is associated with narrowed outflow from either or both ventricles. Among the list of conditions that Hudson (1970) considers possibly contributory to cardiomyopathy are several in which magnesium deficiency or loss have been described: the peripartal state, infantile fibrotic cardiomyopathy, or adult endocardial fibrosis, myofibrosis (or a combination of both), alcoholism, calcium oxalosis, calcific degenerative processes, hyperparathyroidism with myocardial calcification, protein calorie malnutrition, beriberi (Oriental, or the similar, but vitamin-B1-resistant forms seen in alcoholism and in the peripartal period), anemia, severe diarrhea, toxicity from catecholamine or cardiac glycosides, and severe trauma.
Cardiomyopathies occur throughout the world (Shaper, 1968; Editorial, Brit Med J, 1969). In the symposium on "Experimental 'Metabolic' Cardiopathies and Their Relationship to Human Heart Disease" (Ann NY Acad Sci 156, 1969), J. N. Davies (1969) discussed the African cardiomyopathies with and without endocardial lesions, and commented that none of them are confined to the African continent, as is suggested by the name. He suggested that, because coronary arteriosclerosis is so common in the United States, it is possible that many cases of cardiomyopathy are missed. This recalls Caddell's (1965, 1969b) comment that the endomyocardial fibrotic disease in Africa is prevalent in areas where protein calorie malnutrition (associated with magnesium deficiency in the recovery syndrome) is found, and her suggestion that prolonged electrolyte imbalance might be contributory.
T. James (1967) proposed that disease of the small coronary arteries might be an important contributory factor in cardiomyopathies of obscure origin, a position taken also by Varnauskas (1967). James (1967) points out that the clinical manifestations of cardiomyopathy (progressive cardiac enlargement and failure without clear cause, an inordinately high incidence of arrhythmias and conduction disturbances, syncope, sudden unexpected death, and atypical chest pain) can all be due to abnormalities of the small coronary arteries. Considering a basic abnormality to be disease of the cardiac microcirculation permits inclusion of diabetic cardiomyopathy and the cardiomyopathy of progressive muscular dystrophy (James, 1962) in the same category. Neonatal coronary arteriosclerosis-cardiomyopathy complex (Seelig and Haddy, 1976/1980) can be similarly categorized. It further substantiates the premise that loss of myocardial magnesium (and potassium) is likely to be common, not only to experimental but to clinical cardiodegenerative processes (Bajusz, 1965a; Lehr et al., 1966; Lehr, 1969; Lehr et al., 1976/1980), abnormalities of the myocardial microcirculation having been implicated in several of the experimental models that cause both microfocal myocardial necrosis and magnesium loss (Lehr, 1964, 1965, 1966, 1969).
There is no direct evidence that isolated or familial "idiopathic" cardiomyopathy is caused by magnesium loss or deficiency. Ultramicroscopic examination has shown some similarities to those seen in experimental magnesium deficiency and to some of the diseases in which magnesium loss has been described. For example, Hudson (1970) has examined myocardium from hypertrophic cardiomyopathy and shown sarcomeres with widely separated Z bands. Bulloch et al. (1972) found that myocardial biopsy specimens from 12 patients with idiopathic cardiomyopathy were similar to that associated with alcoholism.
8.1.1. Peripartum Cardiomyopathy
The etiology of peripartum heart failure is still a mystery (Editorial, Brit Med J, 1976a), even though it has long been recognized. It was reported in an 1848 textbook by Meigs, and myocardial degeneration was described in women who died in the peurperium by Virchow in 1870. It was reported sporadically in the first third of this century as an important factor in producing heart failure in the peripartal period (Review: Gouley et al., 1937), and is now accepted as definite entity of unknown etiology that is listed as a cause of "primary" cardiomyopathy (Brigden, 1957; Hudson, 1970). The hemodynamic load of pregnancy has been implicated, but in editorial evaluations of this problem, nutritional inadequacy to meet the demands of pregnancy and lactation were considered more likely (Editorials, Brit Med J, 1968, 1976a).
Although magnesium deficiency has not been considered a possible nutritional factor in peripartum cardiomyopathy, there is considerable circumstantial evidence that points to magnesium depletion. Among the conditions associated with peripartal cardiac failure are those that predispose to preeclampsia and eclampsia, with which it is often associated-maternal immaturity, multiple births, and high parity-especially when rapidly successive (Hull and Hafkesbring, 1937; Hull and Hidden, 1938; Teel et al., 1937, Melvin, 1947; Szekely and Snaith, 1947; Walsh et al, 1965; Govan, 1966; Stuart, 1968; J. B. Johnson et al., 1966). These are all conditions that predispose to maternal magnesium depletion. Furthermore the cardiac lesions of peripartum cardiomyopathy (supra vide) strikingly resemble those of experimental "pure" magnesium deficiency
Melvin (1947) and earlier Gouley et al. (1937) and Hull and his colleagues (1937, 1938) commented on the similarity of postpartum heart disease to cardiac beriberi, and implicated probable nutritional inadequacy. However, despite the similarity of manifestations of the disease to Oriental beriberi cardiomyopathy, it is refractory to thiamine therapy (Melvin, 1947; Stuart, 1968). This recalls the refractoriness of alcoholic "beriberi" cardiomyopathy to thiamine and to the dependence of vitamin B1 on magnesium (p. 215). The relatively high frequency of puerperal and "idiopathic" cardiomyopathy in Jamaica, usually in patients with histories of poor nutrition (Walsh et al., 1965; Stuart, 1968), recalls the early demonstration of bovine cardiovascular lesions in Jamaica that were deemed likely to be caused by a "conditioned" magnesium deficiency (Arnold and Fincham, 1950).
Review of the literature shows that the clinical picture is usually one of heart failure, presenting with shortness of breath, palpitations, edema (rarely with acute pulmonary edema), precordial pain, and embolism (Gouley et al., 1937; Teel et al., 1937; Hull et al., 1937; 1938; Szekely and Snaith, 1947; Brigden, 1957; Meadows, 1957; S. Rosen, 1959; Benchimol et al., 1959; Seftel and Susser, 1961; Gilchrist 1963; Walsh et al., 1965; J. B. Johnson et al., 1966; Stuart, 1968; Demakis and Rahimtoola, 1971). Diastolic, and less frequently systolic, hypertension are often found, as is cardiomegaly and abnormal ECGs. Stuart (1968) has commented that close cardiac surveillance may disclose symptomless cardiomegaly or abnormal ECG in an apparently well woman. Toxemia is commonly, but not invariably, part of the history of women who develop peripartal heart failure; Govan (1968) found that cardiorespiratory failure was the cause of fatal eclampsia in his series of 110 cases.
Hypertrophic obstructive cardiomyopathy has been reported by G. Tuner et al. (1968) as an increasingly recognized and often familial form of cardiomyopathy of pregnancy. The Editorial (Brit Med J, 1968) that called attention to this now more common form of cardiac disease of pregnancy suggested that some of the young pregnant women with angina and tachycardia, with ECG abnormalities that persisted after pregnancy (Gilchrist, 1963), might have had this abnormality. This possibility calls to mind the epidemic of supravalvular aortic stenosis syndrome, and other outflow obstructive lesions, that were associated with hyperreactivity to vitamin D at the time of excessive fortification of milk with vitamin D or its use in massive parenteral dosage (Review: Seelig, 1969b) especially in the late 1940s through the 1950s. Is it possible that some of the infants so treated might have been insufficiently hyperreactive to vitamin D to develop the full-blown syndrome, but might have developed silent outflow-obstructive lesions that became overt during the peripartum period? Possibly the presumptively vitamin-D-hyperreactive women might also have had myocardial lesions as a result of the vitamin-D-induced loss of magnesium during infancy and might have been unduly susceptible to both vitamin D and magnesium deficiency during pregnancy
There are several additional fragments of circumstantial evidence suggestive of magnesium deficiency.
1. Patients with this disease have been found to be unusually susceptible to digitalis toxicity, developing multiple premature ventricular contractions that sometimes persist (Walsh et al., 1965; Demakis and Rahimtoola, 1971). (The susceptibility of magnesium-deficient dogs and monkeys to digitalis toxicity should be recalled here.) J. B. Johnson et al. (1966) reported an ultimately fatal case of a 14-year-old mother of twins who, because of her age and the twin pregnancy, had almost certainly been deficient in magnesium. She was unduly sensitive to digitalis.
2. In toxemia, there is commonly aldosteronism and sodium retention (A. Barnes and Quilligan, 1956), and increased catecholamine secretion (Zuspan, 1972), hormones that are secreted in excess in magnesium deficiency and that cause magnesium loss. In addition, women with preeclamptic or eclamptic pregnancies have an exaggerated response to catecholamine infusions (Raab et al., 1956; Zuspan et al., 1964), which have been used as a prognostic test in preeclampsia (Raab, 1957) and to differentiate between essential hypertension and toxemia of pregnancy (Zuspan et al., 1964). The combination of excessive "stress" hormones with probable magnesium deficiency puts cardiomyopathy of pregnancy squarely into the category of "pluricausal" dysionic cardiomyopathy.
3. The susceptibility to peripartal intravascular coagulation, with the risks of damage to the placenta (Bonnar et al., 1971) and of maternal death from embolic phenomena (Arthure, 1968) or that have been difficult to control even with anticoagu1ants (S. Rosen, 1959), might also be related to magnesium deficiency.
4. Among all of the cases reviewed, there was mention of use of magnesium in only two instances: one to measure circulation time, and the other in the management of eclampsia. Decherd and Herrmann (1944) commented briefly that severe tachycardia (of a woman who had had toxemia of pregnancy and developed postpartum heart failure) disappeared after diagnostic intravenous injection of magnesium sulfate. The arrhythmia later recurred and was treated traditionally. In the other instance, magnesium therapy (presumably high dosage) was given to a woman who developed eclampsia and cardiac decompensation during her fourth pregnancy (Teel et al., 1937). The authors noted her "rapid recovery" and lack of recurrence of cardiac manifestations even when she returned, again pregnant, several years later. This was in contrast to five other patients with peripartal cardiomyopathy in this series: Two died and the others required digitalization, two for a short period, one of whom had persistent ankle edema and one of whom had a protracted and incomplete recovery. Whether the magnesium therapy played any role in the complete, rapid recovery of the patient remains speculative. A 38-year-old patient in this series, who died suddenly after she developed cardiac asthma and anasarca during the seventh month of her eighth pregnancy, had myocardial edema, but not necrosis, and slight subendocardial necrosis. She apparently died early in the course of the disease (possibly of arrhythmia), and thus the changes of the small myocardial arteries (intimal hyperplasia and elastica thickening) are of particular interest, since they resemble the changes of experimental magnesium deficiency.
Necropsy examination of patients who died of peripartum cardiomyopathy generally discloses cardiomegaly and dilatation, with focal or diffuse myocardial necrosis and (in later instances) fibrosis, endocardial edema, necrosis and fibrosis and mural thrombi (Gouley et al., 1937; Meadows, 1957; Walsh et al., 1965; J. Johnson et al., 1966; Ledingham et al., 1968; Hudson, 1970; Sakakibara et al., 1970). Several pathologists have reported thickened myocardial arterioles, some times with intimal edema or hyperplasia (Gouley et al. 1937; Teel et al., 1937) and perivascular infiltration around the small coronaries (Meadows, 1957).
Biopsy specimens were examined ultramicroscopically in two reported instances. Perinuclear hydropic vacuolization of myocardial fibers and sarcoplasmic fragmentation was seen 2 months before death from progressive heart failure [7 months after a twin delivery by a 14-year-old girl (J. B. Johnson et al., 1966)]. A 30-year-old woman, who survived the cardiomyopathy that became manifest a week after delivery of her second baby, had widened sarcoplasmic spaces containing irregularly shaped electron-dense deposits, as well as vacuolization (Sakakibara et al., 1970).
8.1.2. Infantile Cardiomyopathy
Coronary and generalized arteriosclerosis of infancy has received more attention in the literature than has infantile cardiac disease (if one excludes the valvular abnormalities and the great vessel and peripheral pulmonary atresias). However, many reporting infantile cardiovascular lesions also mention myocardial and endocardial lesions. Among the lesions tabulated (see Appendix Tables A-5A, A-5B and A-6A, A-6B89.) alone or in combination, are multifocal myocardial necrosis (such as is seen with the small coronary artery damage of magnesium deficiency, subendocardial and papillary muscle necrosis and fibrosis, and endocardial fibroelastosis, as well as massive myocardial infarctions. Among the 157 individual case reports of infants who were stillborn or who died in the first month of life, 37 had myocardial necrosis or cellular infiltration, 23 had myocardial calcinosis, and 38 had myocardial fibrosis. Among the individually cited 253 infants between 1 month and 2 ½ years of age, 72 had necrotic myocardial lesions, 19 had calcific lesions, and 42 had myocardial fibrosis. Endocardial fibroelastosis was reported in 83 of the infants under 1 month of age and in one-third of those of 1 month to 2 ½ years. Over half of the younger group of infants with EFE had outflow obstruction; only about a quarter of those between 1 month and 2 1/2 years of age, tabulated individually, had outflow obstruction. This is in contrast to the surveys of patients selected for EFE, among whom outflow obstruction was found to be very common (Moller et al., 1964; J. Edwards et al., 1965; Oppenheimer and Esterly, 1966). Perhaps a reason for the contrasting findings is the age limitation in cases tabulated and reviewed. Congenital outflow abnormalities-whether the supravalvular aortic stenosis syndrome (SASS), aortic or pulmonic atresia or peripheral pulmonary artery stenoses (alone or in combination)-are also commonly associated with coronary, endocardial, or myocardial diseases and with hyperreactivity to vitamin D (Beuren et al., 1964, 1966; Peterson et al., 1965; Taussig, 1966). Subvalvular aortic stenosis has also recently been suggested as a possible result of hypervitaminosis D (McFarland et al., 1978). There is a relatively small representation of children with outflow abnormalities and cardiofacial peculiarities (which have received much recent attention as familial and isolated cases) in the Appendix tables limited to infants up to 2 ½ years of age. When the endocardial thickening or the arterial disease involves the septum and conducting tissue, arrhythmias and cardiac arrest might result in chronic cardiac disease or in early death. The conditions seen in those surviving beyond infancy include arrhythmias and syncopes. The implication of hypervitaminosis D in such conditions, and the description of calcification of the labyrinth in infants with outflow obstruction, with and without endocardial fibroelastosis and cardiofacies (see cited publications by Beuren et al., 1962, 1964, 1966, in Appendix Table A-6B) raises the question as to whether the syndrome of deaf-mutism, prolongation of the Q-T interval, syncope, and sudden death in children and young adults (Jervell and Lange-Nielsen, 1957) (see cases 187-189, 193, 194, 203, 204, 233: Appendix Table A-6A) might be disorders in which susceptibility to vitamin D toxicity or magnesium loss or malabsorption might play an etiologic role.
In the young infants, prodromal symptoms preceded death by only a few hours to a few days. The symptoms presented are not unlike those reported for infants who died of SIDS. Some of the babies with cardiovascular abnormalities, proved at autopsy, had had signs of illness from the time of birth. ECG tracings typical of ischemic heart disease, were sometimes obtained. Those who had a subacute or chronic course generally were flaccid and quiet, behavior similar to that described by Naeye (1976a) in the SIDS. Those who did not die suddenly or after a short illness of sudden onset generally had had a fairly steady downhill course, with sustained anorexia, vomiting, weight loss, and debility. Several developed hypertension. Coronary arteriosclerosis and focal myocardial necrosis and fibrosis have been found in infants who died suddenly and in others who had been ill with clinically manifest heart disease, many of whose first cardiac manifestations developed at about two to four months of age, the age of peak incidence of SIDS. An international study of 254 cases of sudden unexpected death from cardiovascular disease (in which infants under a year of age were excluded to eliminate the SIDS) found that those who died from 1 to 5 years of age had a disproportionate representation of EFE, pulmonary stenosis, and A-V block (Lambert et al., 1974). Almost a tenth of the total cases were familial. The sudden deaths of the entire series of deaths from 1 to 21 years were associated with myocardial hypoxia in half; about a third had arrhythmias.
Similar total cardiomyopathies, developing postpartum in a mother and in her 7-year-old daughter (Hudson, 1970), raises the possibility that this may have been an instance in which gestational malnutrition (magnesium?) deficiency might have caused maternal and fetal cardiac damage.
The neonatal hypoparathyroidism and hypomagnesemia of infants fed cows' milk might be the human counterpart of the model of cardiorenal necrosis, produced by sodium phosphate loading of parathyroidectomized rats (Lehr et al., 1966; Lehr, 1969). Such infants have a high phosphate/magnesium ratio. Since excesses of both calcium and phosphate (relative to magnesium) are cardiopathic, the prevalence of dietary customs that lead to such imbalances perinatally and in early infancy might be contributory to cardiomyopathy of infants and young children. Persistence of such nutritional imbalances, which might become worse as the intake of high phosphate sodas increases, and as alcohol ingestion begins, can intensify cardiomyopathic lesions that, like the arterial lesions that receive more attention, might have their roots in infancy and possibly even before birth. Since magnesium deficiency causes damage to the intramural small coronary arteries, the perivascular damage to the myocardium that has been reported is not surprising. As in the experimental model, infants who died of cardiovascular disease typically have microfocal myocardial necrosis, infiltration, and fibrosis.
Myocardial mitochondrial and cytoplasmic changes have also been reported. Mitochondria obtained by needle biopsy of a 6-month-old boy with respiratory distress and congestive heart failure had closely stacked, parallel, concentrically arranged cristae, with some cristae filled with electron-dense granular material (Hug and Schubert, 1970). These characteristics are similar to those reported in magnesium-deficient rats (Heggtveit et al., 1964; Heggtveit, 1965b,c). They were not found in the myocardium (at autopsy) of a 6-year-old girl with idiopathic cardiomyopathy, in which there was dissolution of the myofibrillar structures (Hug and Schubert, 1970). Lin (1972) described extensive mitochondrial calcification in the myocardium of a 10-week-old baby boy, who had postductal coarctation, and had had several episodes of cardiac arrest lasting 10 to 50 minutes. The intramitochondrial deposits were needle-shaped dense crystals that resemble those described by Silver and Sordahl (1976/1980) in their in vitro studies of cardiac mitochondria in magnesium-free medium. Lin (1972) noted that ischemia produces intramitochondrial dense bodies that probably represent calcium accumulation, and that the magnesium and potassium contents of ischemia-damaged mitochondria were reduced (Jennings, 1969). An autopsy was obtained 5 hours after the death of a 16-month-old girl who had been in good health until sudden onset of pallor and rapid pulse, with supraventricular tachycardia (320/minute), 18 days antemortem (Haese et al., 1972). The heart showed numerous swollen rounded myocardial cells with partial or complete loss of contractile elements and granular or vacuolated sarcoplasm. Occasional necrotic myocardial cells had adjacent inflammatory cells. The altered cells had many lipid droplets. The mitochondria were distorted. Similar myocardial lesions had been reported in four other female infants (Ross and Belton, 1968; J. Reid et al., 1968; MacMahon, 1971). Of the 13- and 16-month-old baby girls reported by Reid et al.(1968), the first died suddenly while playing, with no prior evidence of illness. The second was admitted with a history of vomiting, drowsiness, and left hemiplegia after a fall. She was found to have right bundle branch block and supraventricular tachycardia. She was unresponsive to therapy, developed new thrombotic events, and died 3 days after admission. Reid et al. (1968) considered the abnormal cells in the myocardium and in the region of the atrioventricular node as a probable reaction to degenerating myocardial fibers. The 13-month-old girl reported by MacMahon (1971) was the seventh child; the preceding sibling had had multiple developmental anomalies, including cardiac disease, and died at 16 months of age. The propositus had been well until 15 hours before admission. Repeated episodes of vomiting and then tachycardia led to hospitalization; ECU showed arrhythmia and a rate of 200/minute. Half an hour after digitalization and starting intravenous fluids, ventricular fibrillation developed. Recurrent episodes were treated by external cardiac massage, defibrillation, and finally adrenalin, calcium chloride, and isoproterenol. The next day she developed tonic-clonic seizures. She died 62 hours after admission, and at autopsy had many "xanthoma cells" throughout the myocardium, in the subendocardium and in the septum, involving the conducting system. No data were given as to the intervals between the births of the patient and her six siblings, but the multiple anomalies of the immediately preceding baby suggest that the mother might have been nutritionally depleted, possibly of magnesium. Thus, her last infant might also have been low in magnesium stores, and might have had small coronary arterial disease such as has been implicated in conduction tissue disease.
Another baby girl (8½ months old) first developed an episode of paroxysmal atrial tachycardia (PAT) that responded to digitalis about 2 months before her death Bove and Schwartz, 1973). The PAT recurred 3 days before her death (while she was still on digitalis), and she was treated with direct current shock and pacing, to which she was unresponsive, developing profound hypotension necessitating administration of epinephrine and isoproterenol. Necropsy examination showed microfoci of acute ischemic necrosis and cells resembling storage histiocytes, containing lipid, scattered throughout the left ventricular wall, the interventricular septum, and both atria. Ultramicroscopy showed mitochondria, many of which were swollen and contained amorphous dense inclusions. In focal areas the cristae were stacked; the outer membranes of adjacent mitochondria were fused to form electron-dense segments. There were focal aggregates of swollen lipid-laden myocardial fibers and myofibrillar membrane-limited dense granular that seemed to be spatially related to early Z-band degeneration. These findings resemble those described under magnesium deficiency. Possibly, the lipid accumulation in this and the preceding case might have been contributed to by the catecholamines given in an effort to correct the hypotension. It is conceivable that the refractory hypotension of these infants might have been the result of magnesium depletion; in magnesium deficiency, in vitro, arterial smooth muscle exhibits markedly diminished arterial contraction in response to vasoactive amines (pages 179-183).
Alcoholic cardiomyopathy has been considered a nutritional disease, caused predominantly by thiamine deficiency and by deficiencies of other vitamins (Blankenhorn, 1945). There was then a shift in emphasis, implicating a directly cardiotoxic effect of alcohol, since thiamine is not therapeutic in a substantial number of chronic consumers of hard liquor, and many of the patients are well nourished and respond to prolonged bed rest and abstinence from alcohol (Burch and DePasquale, 1969). More recent work focuses attention on the nutritional aspect of the disease, but this time with the major emphasis on magnesium deficiency as a common denominator in the failure to respond to thiamine, in the arrhythmias seen in alcoholic cardiomyopathy, and in the cardiac lipid accumulation and ultramicroscopic changes.
Thiamine loses enzymatic activity in magnesium-deficient rats, which exhibit signs of thiamine deficiency unless magnesium is repleted (Zieve et al., 1968a,b; Zieve, 1969). Furthermore, thiamine levels have been shown to fall in liver and kidneys of magnesium-deficient rats (Itokawa et al., 1974c). Magnesium-deficient alcoholics are unresponsive to vitamin B (and other B vitamins) until their magnesium is repleted (Zieve, 1975). Magnesium deficiency has long been recognized in alcoholism (Flink et al., 1954; Review: Rink, 1976/1980); it can be secondary to low intake, malabsorption, and, if cirrhosis develops, secondary aldosteronism (Review: Massry and Coburn, 1973). The dependence of thiamine activity on magnesium as a co-factor is relevant (not only to the psychoneurologic manifestations of alcohol withdrawal) but to at least three of the metabolic aberrations that affect the heart in alcoholism.
1. Itokawa et al. (1973) have found that there is increased lipogenesis, both in magnesium-deficient and in thiamine-deficient rats. They demonstrated increased lipid and cholesterol in liver and kidneys and hypothesize that these deficiencies lead to a general increase in lipid synthesis, possibly by blocking the pathway of acetate to the tricarboxylic cycle, shunting the acetate to the lipogenesis pathway. Thus, it is possible that the magnesium-thiamine deficiencies are contributory to the accumulation of myocardial lipid droplets that have long been recognized as characteristic of alcoholic cardiomyopathy. Thiamine deficiency has also been shown to cause myocardial catecholamine accumulation (Raab and Supplee, 1944), an effect also demonstrated for magnesium deficiency.
2. Another metabolic aberration to which magnesium-thiamine deficiency might contribute is acetaldehyde accumulation, which might result from blockage of the thiamine-dependent step by which acetaldehyde goes to pyruvate (Altman and Dittmer, 1968). Discussed elsewhere in this volume is the acetaldehyde-induced arrhythmia (which has been shown to be protected against by β-adrenergic blockade), suggesting that it is mediated by catecholamine release, but that might just as well be mediated, in alcoholism, by magnesium deficiency. Contributory to the presumed increased catecholamine effect might be ethanol's interference with catecholamine metabolism (V. Davis et al., 1967b).
3. There is an interrelationship between magnesium deficiency and thiamine (excess) that bears on serotonin levels and metabolism (Itokawa et al., 1972b). Magnesium (as the EGTA chelate) inhibits the release of serotonin from platelets (Henson, 1969). Magnesium deficiency, particularly in the presence of excess thiamine, inhibits the oxidation of serotonin (Itokawa et al., 1974a). Whether ethanol's interference with serotonin's metabolism (V. Davis et al., 1967a) might be intensified by magnesium deficiency and thiamine therapy might be worth investigating.
Further inferential evidence that magnesium deficiency is contributory to alcoholic cardiomyopathy derives from study of the cardiac lesions. (Heggtveit 1965a), who had observed that the coronary arterioles of magnesium-deficient rats were edematous, also observed that intravenous infusion of 20% ethanol into rats caused significant swelling of the capillary endothelial cells (Heggtveit and Nadkarni, 1971).
Pintar et al. (1965) reported edema and disorganization of the layers of the coronary arterioles, with perivascular foci of edema, necrosis, and spotty calcification in the hearts of three alcoholic men, 53 to 63 years of age. One also had subendocardial fibrosis. Their aortas and major branches of their coronary arteries showed only minimal atheromatous changes. Alcoholics who died with early stages of cardiomyopathy were reported to have edema of the coronary vessels (Benchimol and Schlesinger, 1953). Three patients with advanced alcoholic cardiomyopathy, who had died after developing clear signs of acute transmural infarction, had a coronary obstruction but had periarterial myocardial fibrosis (Regan et al., 1975). These investigators speculated that the fibrosis around the myocardial coronary arteries might have interfered with their ability to dilate, thereby causing confluent necrosis when oxygen requirements increased.
Pintar et al. (1965) suggested that the vessel wall edema of their alcoholic cardiomyopathic patients might have resulted from hypomagnesemia. Recently, Hungerford and Bernick (1976/ 1980) gave histologic details of the coronary arterial structural disorganization caused by magnesium deficiency.
Ultramicroscopic studies of alcoholic cardiomyopathic hearts and hearts from magnesium-deficient animals also show similarities. Heggtveit and Nadkarni (1971) reported significant swelling of the mitochondria and sarcoplasmic reticulum after an acute ethanol load in rats, but were unable to induce cardiomyopathy by long term alcohol feeding. Szanto et al. (1967) however, did find some changes in the mitochondria and sarcoplasmic reticulum of alcohol-fed rats. Mice seem to be more susceptible to alcohol cardiomyopathy. Sohal and Burch (1969) found strikingly separated intercalated discs of mice given water containing 15% ethanol for three weeks. Burch et al. (1971) then reported that alcohol (ethanol, beer, or wine) produced myocardial damage in mice even when they are otherwise properly fed. The mice developed swelling of the mitochondrial cristae and of the sarcoplasmic reticulum, disorientation of the myofibrils, expansion of the intercalated disc, and accumulation of fatty deposits and dense particles. These changes are very like those reported by Hibbs et al. (1965) in six autopsied cases of alcoholic cardiomyopathy: severe mitochondrial swelling, degeneration and fragmentation of the cristae, and formation of dense inclusions. There was also pronounced swelling of the sarcoplasmic reticulum, excessive lipid accumulation, and myofibrillar degeneration and lysis.
Needle biopsy specimens from patients with alcoholic cardiomyopathy also showed severe mitochondrial changes, with subsequent derangement and fragmentation of contractile elements (Alexander, 1966b). This investigator commented that the ultramicroscopic changes resemble those of magnesium-deficient animals, and suggested that the myocardial lesions might be secondary to ethanol-induced magnesium deficiency, rather than a direct consequence of the alcohol per se. He referred to the evidence that the metabolism of the vasoactive amines, catecholamine and serotonin, is interfered with by ethanol, and considered the possibility that their accumulation in cardiovascular tissue of alcoholics might thereby be enhanced.
Myocardial biopsy specimens from eight patients with alcoholic cardiomyopathy, and from twelve with idiopathic cardiomyopathy were compared by Bulloch et al. (1972). The major ultrastructural lesion in both was contractile element-sarcoplasmic reticulum disorganization. Swelling of the sarcoplasmic reticulum was early and generalized in the alcoholic cardiac disease; it was focal and inconstant in idiopathic cardiomyopathy. In this series of cases, mitochondrial damage was not a major lesion in either diseases.
The similarity of the ultramicroscopic findings in these two diseases of such diverse etiologies, and their similarities to the changes of magnesium deficiency and of a variety of experimental models characterized by loss of myocardial magnesium suggest that testing for tissue losses of magnesium, and trial of magnesium supplementation be investigated.
8.1.4. Diabetic Cardiomyopathy
Diabetes mellitus is one of the diseases that was first recognized to be associated with magnesium deficiency (Martin and Wertman 1947; Martin et al., 1951 1958; Martin, 1969, Jackson and Meier, 1968). Additionally, diabetics commonly have diffuse endarterial proliferative small vessel disease (Ditzel, 1954) that resembles the arteriolar lesions of magnesium deficiency. In a study of small vessel disease of the diabetic heart, Rubler et al., (1972) presented cases with fibrosis throughout the myocardium, in conjunction with the damaged intramural vessels Among 73 patients with "idiopathic" primary cardiac disease, studied by Hamby et al. (1974) 16 had diabetes mellitus, a high frequency of diabetes that was statistically significant. Autopsies were performed in 3 of the 4 diabetic patients with cardiomyopathy who died; all 3 had small coronary, but not large coronary artery disease. In this series of cases, only one of 28 patients with cardiomyopathy without diabetes mellitus had small coronary vessel disease.
T. James (1967) suggested that arrhythmias of diabetes mellitus might be caused by small coronary arterial disease of the vessels supplying the conducting tissue of the heart. Impaired atrioventricular conduction has been found significantly more frequently in patients with diabetes mellitus or abnormal glucose levels (Rubler et al., 1975) than in patients with other diseases.
Part II: Chapter 9
MAGNESIUM DEFICIENCY IN THE PATHOGENESIS OF CARDIOVASCULAR DISEASES
In the early subacute magnesium-deficiency study of Kruse et al. (1932), convulsions were produced in 86% of the rats by the 18th day, with death occurring after one or more convulsions in 93%. Tachycardia was manifest during the preconvulsive period, and bradycardia with marked arrhythmia just before the convulsions started. Greenberg and Tufts (1938) confirmed these findings, and showed additionally that ECGs, taken while the rats were unconscious from the convulsive seizures, revealed a sinoauricular block, with occasional skipped and ventricular beats. Of 10 rats with less severe magnesium deficiency, such that despite manifest nervousness only one developed convulsions, seven survived long enough to have ECGs recorded the day before sacrifice on day 62. These rats exhibited little change in heart rates (which were slightly slower than were those of control rats on the same diet to which magnesium had been added) but had lengthened P-R intervals. Five of the seven surviving deficient rats had additional ECG abnormalities: Three had numerous extrasystoles, two had abnormally high takeoff of the ST segment in lead III, one with partial heart block and one with auricular extrasystoles.
Production of magnesium deficiency (average serum magnesium = 0.4 mEq/liter) in young dogs, with a diet similar to that used by Kruse et al. (1932), produced no significant difference from control heart rate (Syllm-Rapoport et al., 1962). There was a highly significant shortening of the atrioventricular conduction time (P-Q interval) and of the intraventricular conduction time (QRS in Lead II). There was some prolongation of the electrical systole (QT interval). There was an increased incidence of negative T waves in leads I, II, and III that was statistically significant in lead III. The voltage of the negative T waves in leads I, II, and III was statistically significant in lead III. The voltage of the negative T waves in leads I and II was almost half that of controls. Striking inversion of the T waves was seen in several of the deficient animals. Comparably severe magnesium deficiency, produced with a semisynthetic diet, and that produced severe hypomagnesemia (< 0.5 mEq/liter), but no significant effect on serum potassium or calcium), and that caused arterial and multifocal myocardial lesions, was also associated with ECG abnormalities (Wener et al., 1964). These dogs developed sinus tachycardia, but little difference from control PR, QT, or QRS intervals. There was frequent occurrence, as in the previous group of dogs, of T-wave abnormalities: flattened or inverted T waves, especially in leads III, aVL, and V. They also had consistent RST-segment depression.
Subacute magnesium deficiency for three months in puppies that resulted in irritability and occasional convulsions also resulted in marked sinus tachycardia, peaking of the T waves, and ST-segment depression (Vitale et al., 1961). These dogs also became more susceptible to digitalis toxicity. These investigators pointed out the relationship of these ECG changes to the magnesium-induced shift in potassium. Although the magnesium-deficient dogs also developed hypokalemia, Vitale et al. (1961) referred to the loss of intracellular potassium that results from magnesium-deficient interference with mitochondrial enzymatic activity. They speculated that there might be a relatively greater decrease in intracellular than plasma potassium with a relative hyperkalemia. In support of this premise, was the peaked T wave of their magnesium-deficient dogs that resembled that seen in hyperkalemia. Ono (1962) confirmed these findings with young dogs maintained for four months on the same diet as used by Vitale and his co-workers (1961). He, too, found peaking of the T waves, especially in lead VR, when the serum magnesium levels decreased to 0.7 mEq/liter, with concomitant falls in serum potassium. Depression of the ST segment in limb or chest lead appeared with serum magnesium levels below 0.8 mEq/liter. The P-R interval increased slightly as the hypokalemia worsened. The Q-T interval remained almost normal. There were also occasional premature contractions. Comparable changes were produced by magnesium deficiency in monkeys (Vitale et al., 1963), except for bradycardia and elevated ST segment in severely deficient monkeys. The peaking of the T waves and the ST segment changes were comparable to those seen in hyperkalemia, even though the animals had hypokalemia. This group then tested their original postulate that there might be a local relative hyperkalemia (of the extracellular/intracellular potassium concentration) in the magnesium-deficient heart (Seta et al., 1965, 1966). Rats fed diets low in both magnesium and potassium had substantial reductions in myocardial potassium and magnesium levels. Rats on a low magnesium, adequate potassium intake had almost normal serum potassium level, but markedly subnormal myocardial potassium (Seta et al., 1965), supporting the premise that the hyperkalemia-like ECG of relatively early magnesium deficiency reflects local relative hyperkalemia. Electrocardiographic changes were observed at two-week intervals: T-wave peaking developed within two weeks of instituting the magnesium-deficient diet (best seen in the left precordial unipolar lead). QRS widening and tachycardia were additional early changes (Seta et al., 1966). ST segment depression, ventricular premature beats, and bigeminal rhythm were also seen in some of the dogs. The ECG changes of dogs deficient in both magnesium and potassium resembled those of potassium deficiency, but the terminal T-wave inversion was more marked. The P-R interval and the QRS segment were prolonged, and there was slight ST-segment depression. The heart rate, unlike that of the animals that were deficient in magnesium but not in potassium, did not change. Dogs that were kept on the magnesium-deficient diet for nine months developed an ECG pattern indistinguishable from that of doubly deficient dogs after two months (Seta et al., 1966)
Electrocardiographic changes, like those of subacute experimental magnesium deficiency have been reported from studies of cattle pastured on land low in available magnesium (Willers et al., 1965). The ECG criteria for detection of the disease termed "bovine arteriosclerosis" are tented T waves, prolonged QRS interval, and elevated ST segments. The investigators noted that such events in man are associated with hyperkalemia, endocardial thickening, and conduction system-interference. Autopsy reports (of cattle from the herd tested electrocardiographically) showed that endocardial thickening and coronary calcific arteriosclerosis were characteristic (Lynd et al., 1965). Larvor et al.(1964a) found that magnesium-deficient calves had tachycardia and shortened PQ intervals. One calf that developed myocardial degenerative changes had had a diphastic T wave. Reference to the conduction disturbance recalls the early microscopic study of magnesium-deficient calves that showed not only endocardial plaques and fibroelastosis and myocardial necrosis, but lesions of the Purkinje fibers (Moore et al., 1936; 1938; Arnold and Fincham, 1950). It also recalls the evidence that the interventricular septum has the greatest avidity for magnesium (Glaser and Brant, 1959; Glaser and Gibbs, 1962; Lazzara et al., 1963; Burch et al., 1965). Thus, the dysrhythmias of magnesium deficiency probably reflect high magnesium requirements of the conduction system, and secondary potassium shifts out of the myocardial cells.
Acute sudden magnesium depletion by hemodialysis has not produced as significant ECG alterations as have subacute or chronic deficiencies. Danzig and Walker (1955) depleted dogs of magnesium over a six-hour period by dialysis, using a magnesium-free, but otherwise physiologically constituted dialysate. The ECGs s the end of dialysis, when the plasma magnesium was 0.34-0.70 mEq/liter showed an increased heart rate and decreased QT interval. Comparable reduction in plasma magnesium during dialysis for 2 1/2 hours caused only 15% increase in rate and slight decrease in PR and QT intervals (Grantham et al., 1960)
Baby pigs, on a synthetic milk diet that was severely deficient in magnesium, developed bradycardia, increased R and T wave potentials, and inverted T waves in the standard leads (Miller et al., 1964a). Moderate magnesium deficiency resulted in tachycardia with a normal R-wave potential. Acute calcium deficiency also produced bradycardia, with a lengthened ST interval.
Bajpai et al. (1978) have correlated the ECG changes produced by hypomagnesemia in rats with abnormalities in mitochondrial oxidative phosphorylation. They confirmed the significant reduction of the P-, QRS-, and T-wave voltages of magnesium deficiency, and attributed the changes to decreased energy production associated with the decreased oxidative phosphorylation. They propose that magnesium deficiency reduces the amount of current transmitted from cell to cell, as a result of increased resistance in the intercellular connections (desmosomes) as these membrane structures swell [similarly to the swelling of the plasma membranes of magnesium deficient erythrocytes (Elin, 1978)].
It might be that the severe forms of magnesium deficiency that are associated with bradycardia and depressed, prolonged ST segments might reflect concomitant hypocalcemia and hypokalemia (Fig 9-1B; Seelig 1969a). The ECGs of the less severe, subacute, or chronic magnesium deficiencies resemble not only that of hyperkalemia but also that of hypercalcemia, or a combination thereof (Fig. 9-lA). Thus, the magnesium-deficiency ECGs reflect also the concomitant or resultant abnormalities of the other two cations that affect the cardiac conduction system, and not the magnesium status alone.
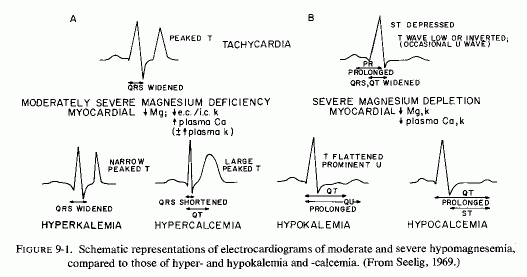{kind=link}
It is provocative that the ECG of magnesium deficiency also resembles that seen in myocardial ischemia of coronary insufficiency: flattened, inverted, or peaked T waves and ST depression, as well as abnormally long QT interval. Its ST depression and T-wave inversion also resemble the ECG of subendocardial infarction, an interesting point, since the small intramural coronaries have been shown to be most compromised in magnesium-deficient animals, and the subendocardial area is most susceptible to ischemia under such circumstances. The presence of endocardial fibroelastosis in infants with perinatal factors that increase the risk of hypomagnesemia is further suggestive evidence that magnesium deficiency might be contributory to the infantile cardiovascular diseases discussed earlier, and that its loss from the heart might be a factor in the ischemic ECG.
Basic to the role of magnesium in maintaining or restoring normal cardiac rhythmicity and in preventing hyperexcitability is its role in maintaining intracellular accumulation of potassium against a concentration gradient, and in counteracting excess calcium influx.
DeCarvalho (1965) has reviewed the factors controlling electrical activity in heart muscle, which is strongly dependent on the electrolyte balance between the cell and its environment. Potassium ions are particularly important in the genesis of cardiac transmembrane potentials, and affect the cardiac rate, impulse conduction, excitability threshold and refractoriness. He considered the influence of alterations in extracellular potassium concentrations on the polarity of the myocardial cell and on transmission of impulses in the special conducting system of the heart. High concentrations of extracellular potassium depress impulse propagation in the atrium and in the His-Purkinje system; low extracellular concentrations depress the atrioventricular (A-V) nodal area, A-V block ensuing with very low (1.5 mM) extracellular potassium levels. Under normal circumstances, potassium is extruded from the myocardial cell during systole. Its return during diastole is an energy-dependent process, since it entails transport against a concentration gradient (Raab, 1969).
The active transport of potassium into, and sodium out of the myocardial cell is dependent on the integrity of the mitochondrial enzyme system. Baltscheffsky 1956, 1957) first postulated that magnesium plays a specific role in the respiratory control of the mitochondrion, since it is a cofactor in the oxidative phosphorylation reactions. Without magnesium, the respiratory rate decreases: there is "uncoupling" of oxidative phosphorylation. He also suggested that magnesium is essential to mitochondrial integrity. A. Schwartz (1971/1972) diagrammed the structure and functions of the mitochondrion which contain magnesium-dependent enzyme systems of the Krebs cycle (and provide most of the energy requirements, as well as those controlling oxidative phosphorylation). Lehninger (1962) showed that the mitochondrial functions are responsible for electrolyte and water transport. Aikawa 1965) reviewed the data on the enzymatic importance of mitochondria and hypothesized that magnesium is essential for the metabolic activity of all subcellular particles. He speculated that there might be an "unknown carrier molecule" that might be involved in the active transport of the magnesium ion across the inner membrane of the mitochondrion, such as has been identified in cardiac mitochondria (Blondin, 1975; Green et al., 1975).
The importance of oxidative phosphorylation, particularly in Mg-activated membrane ATPase (that is vital in electrolyte transport), was first shown in noncardiac tissue such as nerves, brain, kidney, and erythrocyte membranes (Skou, 1957, 1960, 1962; Post et al., 1960; Dunham and Glynn, 1961; Whang and Welt, 1963; Welt and Tostesen, 1964; Welt, 1964), and in cardiac and other mitochondria and microsomes (Nakamura et al., 1961; DiGiorgio et al., 1962; Auditore, 1962; Auditore and Murray, 1963; Vitale et al., 1963; Schwartz, 1962; Schwartz and Laseter, 1964).
The loss of myocardial potassium in magnesium-deficient animals was first attributed by Vitale and his colleagues (1975a,b) to the uncoupling of oxidative phosphorylation. The myocardial cells (which are vulnerable to magnesium loss because of the high percentage exchangeability of their magnesium, not only lose the capacity to accumulate potassium against a concentration gradient and pump out sodium, but show concomitant mitochondrial disorganization, a not surprising correlation in view of the dependence on the integrity of the mitochondrial enzymes systems for active electrolyte transport.
Epinephrine, whether injected (W. Robertson and Peyser, 1951) or secreted as a result of stress (Raabet al., 1968), causes a decrease in the intracellular potassium/sodium ratio. Using the potent β-adrenergic agonist (isoproterenol), Lehr et al. (1966) showed that the earliest myocardial electrolyte changes were significant decreases in magnesium and phosphorus and an increase in calcium. These changes were noted at 3 hours after injection of the catecholamine, in association with mild microscopic evidence of necrosis, but no significant changes in potassium or sodium. There was a significant rise in myocardial sodium and a minor fall in myocardial potassium at 12 hours, at which time the abnormalities in magnesium, calcium, and phosphorus were greater and all of the rats had severe myocardial necrosis.
These observations may help to explain the marked similarity of the ECG produced by excessive sympathomimetic substances (Raab, l943b) and those produced by nutritional magnesium depletion, whether produced in the experimental animal or as a result of chronic alcoholism or protein calorie malnutrition (Seelig, l969a). In both there can be elevation or depression of the ST segment, abnormalities of the T wave, ranging from a high pointed shape to inversion and prolongation of the QRS or QT interval. Raab (1943b) pointed out that the epinephrine-ECG pattern reflects the relative hypoxia produced as oxygen consumption exceeds oxygen supply. It is of interest that chronic magnesium deficiency has been shown to cause luminal narrowing of intramyocardial coronary arteries and also to interfere with normal mitochondrial respiration supra vide).
The DOCA saline pretreated rats that showed myocardial magnesium depletion and that died of ventricular fibrillation 15 to 30 minutes after minimal doses (60-100 µg/kg) of isoproterenol, developed auricular and ventricular arrhythmias, progressing to fibrillation as the β-agonist dosage was raised. Epinephrine, as α- and β-agonist, elicited arrhythmias and ventricular fibrillation less consistently, and only when α-adrenergic receptors were blocked (Guideri et al., 1974).
It is surprising that the ECG changes and myocardial damage produced in rabbits stressed by being kept in a vertical position were significantly protected against by oral administration of magnesium chloride (1 g/kg) twice daily, whereas those injected with epinephrine (0.2 mg/kg) intravenously were not similarly protected by MgCl2 (Pokk, 1971/1973). The catecholamine-injected rabbits had been given an unspecified amount of the magnesium every five days before the injection and thereafter. Perhaps the amount given was insufficient to achieve the amelioration of ECG and myocardial changes that was seen in the stressed, magnesium-dosed rabbits. The ECG changes included bradycardia, large R waves, and depressed ST segments.
9.2.3. Postinfarction/Catecholamine/Free Fatty Acid/MagnesiumInterrelationships with Arrhythmia
It has been shown that blood and urine catecholamine levels are increased in patients who are severely ill after a heart attack and the catecholamines have been implicated in the postinfarction arrhythmias (Gazes et al., 1959; Richardson et al., 1960; Valori et al., 1967; McDonald et al., 1969; Editorial, Lancet, 1969a). High levels of circulating free fatty acids have also been implicated in postinfarction arrhythmias (Kurien and Oliver, 1966; Kurien et al., 1969, 1971), and the two findings have been correlated by some, in view of the catecholamines' lipolytic effects McDona1d et al., 1969; Editorial, Lancet, 1969b)
When corn oil was added to the diet of sodium phosphate mineralocorticoid treated (ESCN) rat, it developed infarctlike myocardial lesions and electrocardiographic abnormalities that were similar to those produced by magnesium deficiency in association with local relative hyperkalemia (Vitale et al., 1961, 1963; Seta et al., 1966): There was prolongation of the PR and QRS segments, low voltage, and peaking of the T wave, with atrial fibrillation and conduction abnormalities that developed at about the time that necrosis became visible (Varga et al., 1970). Amiloride protected against the severe ECG changes, but tachycardia persisted and the amplitude of the PR and S waves remained elevated. Cardiac necrosis was almost completely prevented. Serum and myocardial electrolyte analyses of rats sacrificed on the fifth day of study suggest that in this model, the amiloride protection might have been mediated by protection against myocardial necrosis closely resembling those of his electrolyte-steroid cardiac-necrosis (ESCN) experimental model in that all produce extensive, usually multifocal myocardial necrosis. Excessive concentrations of epinephrinelike substance in the heart of a young athlete who had died suddenly (Raab, 1943a), and in hearts of patients who had died with angina pectoris and other cardiac dysfunctions (Raab, 1943b), and the similarity of the ECG changes of patients with IHD to those of animals or humans given epinephrine, led Raab to consider stress-induced hormonal (catecholamine and corticosteroid) excess as basic to the disorder he termed cardiac "dysionism." He observed that major shifts in myocardial electrolytes can lead to disturbances in cardiac rhythm, contractility, structure, and ultimately to cell necrosis. His emphasis was on the depletion of intracellular potassium, but he observed that this was usually paralleled by loss of glycogen and magnesium and by entry of sodium into the myocardial cells.
When cardiac function is inadequate for the load capacity of the heart, relative ischemia develops, which can be manifested by angina, sudden death from arrhythmia, or congestive failure. Hochrein and Lossnitzer (1969) have pointed out that when there is hypoxic cardiac dysfunction or failure, the myocardial metabolism is characterized by shift toward anaerobic from aerobic metabolism, with loss of magnesium and potassium and gain of sodium chloride and water (Fig. 9-2A), with resultant myocardial edema and decreased energy production for the amount of oxygen consumed. A similar pattern results from cardiac overload, except that there is first increased lactate consumption with intensified glycolysis, and subsequent inhibition of glycolytic metabolism, again with loss of myocardial magnesium and potassium (Fig. 9-2B).
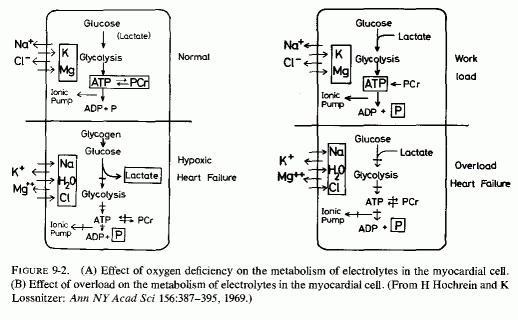{kind=link}
On the other hand, anoxia itself causes loss of magnesium from the myocardium, as well as increased myocardial lipid accumulation (Review, Opie, 1968). which can further decrease the available magnesium. Even venous occlusion of the arm with a blood pressure cuff causes egress of magnesium from the cells, as reflected by immediate rise in local serum magnesium levels (Whang and Wagner. 1966; Nielsen, 1969). Thus, the similarities between the ECGs of coronary insufficiency and of magnesium deficiency are not surprising. The changes that occur with treatment of the cardiac disease, particularly that increase myocardial calcium and decrease myocardial magnesium (i.e., cardiotonics) and that intensify potassium loss (i.e., diuretics), to which magnesium deficiency contributes (Review: Seelig. 1972) can account for the wide variety of ECG changes seen with cardiac ischemia and decompensation and at different stages of magnesium deficiency.
The use of acid citrate dextrose (ACD) solution for preserving blood without coagulation is known to remove ionized calcium, and thus calcium is usually added. However, its equal binding capacity for magnesium has not been as widely appreciated, with resultant production of arrhythmias during exchange transfusion in infancy and in open-heart surgery. Killen et al. (1971) have shown that total and ionized magnesium levels dropped to below 1.5 mEq/liter and to about 0.5 mEq/liter, respectively, within two hours of the infusion in dogs. Heparin has been recommended as an anticoagulant, to avoid this problem, but Romero et al. (1973) performed cardiopulmonary bypass using heparinized blood in dogs, and found drop of serum magnesium from a pre-bypass level of 1.6 to 1.2 mEq/liter, which was sustained for the two hours of the bypass and for the hour of observation thereafter. Thus, to avoid the arrhythmias of exchange transfusion and of open- heart surgery, addition of magnesium to the prime is recommended. That the optimal magnesium concentration in the infusate or blood prime might be considerably higher than the physiological concentration, as suggested by the few who have written reports recommending the clinical use of magnesium during open-heart surgery, is indicated by the study of Hearse et al. (1978). Using a rat heart model of cardiopulmonary bypass, they showed magnesium to be the single most effective component of any infusate tested. The concentration at which maximal protective activity was achieved was 15 mmol/liter. Increasing the magnesium concentration from 0 to 15 mmol/liter produced a progressive and significant improvement in the recovery of function during the reperfusion. There was a striking increase in protection between 0 and 2.4 mmol/liter and another at 15 mmol/liter; thereafter the protective effect declined with increasing magnesium concentrations (Table 9-1).
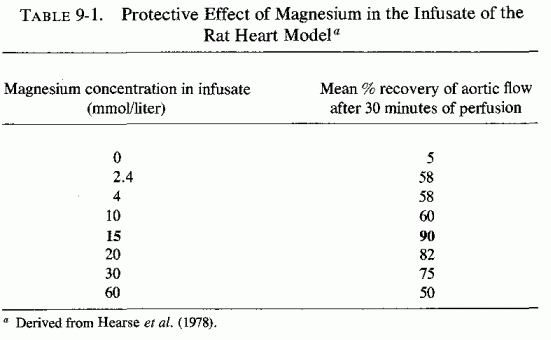{kind=link}
ECG abnormalities (similar to those seen in magnesium-deficient animals or in animals subjected to stress or given hormonal, drug, and electrolyte challenges that produce loss of myocardial magnesium) have been seen in several clinical conditions that have caused magnesium depletion. Many conditions have been listed as associated with hypokalemic or hypocalcemic ECGs, or both (Surawicz and Lepeschkin, 1953; Judge, 1968; Fletcher et al., 1967; Table 9-2). It is of interest that all of these conditions have been associated with magnesium deficiency: hypomagnesemia, low tissue levels, both, or low tissue levels but high serum levels. Electrocardiograms have been recorded while volunteers were depleted of magnesium. Among the diseases in which abnormal ECGs have been recorded, the most acute situations, those that produce sudden and severe hypomagnesemia, often in association with preceding or concurrent stress, are those during which citrated blood is used. Exchange transfusions of infants, open heart surgery, and extensive surgical procedures or repair of blood loss secondary to serious trauma, are the most obvious examples. The first recognition of the risk of hypomagnesemia and associated arrhythmia in patients receiving long-term intravenous infusions was reported by Flink et al. in 1954. The following years this group called attention to the hypomagnesemia of alcohol abuse and diuretic overuse (Flink, 1956; McCollister et al. 1958), both agents that have been associated, as well, with arrhythmias. Infants with "primary" or "idiopathic" myocardial diseases have also been found to have ECG abnormalities resembling those of magnesium deficiency.
{kind=link}
Two normal men were fed diets low in magnesium (1-2.5 mM/day; or 2-5 mEq/day or 25-60 mg/day) and high in calcium for 39 and 48 days, respectively, and were given intravenous infusions of sodium and potassium sulfate to augment renal magnesium loss. One, who developed hypokalemic alkalosis on day 46, developed a hypokalemic ECG, despite a potassium intake of more than 40 mM/day. The plasma potassium rose and the ECG reverted to normal during magnesium repletion (Dunn and Walser, 1966). Seven patients who had had radical head and neck surgery for carcinoma, and thus could be kept on a controlled magnesium-deficient liquid diet for prolonged periods, were more severely depleted of magnesium (Shils, 1969a). Their daily magnesium intakes were 0.5 to 0.8 mEq (60 to 10 mg) for 42 to 266 days. Serial ECGs were obtained on all subjects. Three, who had been depleted for 42, 104, and 117 days, developed changes in the T waves consisting of broadening and decreased amplitude (or occasionally inversion), U waves, and slight prolongation of the QT interval. Two of these patients also had decreased voltage and one had some shortening of the ST segment. A fourth patient had a prolonged QT interval. These changes were associated with low levels of magnesium, calcium and potassium. Only one patient with severe electrolyte changes had no ECG abnormality. The two patients with the least disturbance in electrolytes had no significant ECG changes. It is noteworthy that during the early period of magnesium repletion, two of the patients' low serum potassium and calcium persisted even though their serum magnesium levels had become normal. Their ECGs remained abnormal until later in magnesium-repletion period, at a time coinciding with restoration of normal serum calcium and potassium levels. Possibly this reflects correction of the ECG when the body stores (including cardiac levels) of magnesium were sufficiently repleted to permit restoration of normal mitochondrial and parathyroid function, without which inability to maintain normal potassium and calcium levels is not surprising, despite their supplementation.
9.3.2.1. Exchange Transfusion
The evidence that exchange transfusion with ACD blood has produced acute magnesium deficiency in newborn infants has been discussed earlier. The most consistent change associated with a drop in serum ionized magnesium to below 0.8 mEq/liter, was characterized by a flat T wave (Bajpai et al., 1971, 1972). A similar tracing was seen in another infant, who was unsuccessfully treated with calcium gluconate for hypocalcemic seizures, noted first six days after he had undergone an exchange transfusion (Dooling and Stern, 1967). A flat T wave was noted in the third week of life when, despite continuation of calcium therapy and sodium bicarbonate treatment of his acidosis, the baby continued to suffer disastrous seizures, which were finally attributed to his concomitant hypomagnesemia (0.6 mEq/liter), reported first from blood taken on the 11th day of life. His tremulousness and seizures ceased in response to 0.25 ml of 50% magnesium sulfate every six hours (providing 25 mEq in 24 hours), but his serum magnesium remained low (0.76 mEq/ liter); it was at this time (three days after the magnesium therapy had been started) that the abnormal ECG was first observed. The infant required 0.5 ml of 50% magnesium sulfate intramuscularly every eight hours to bring his serum magnesium up to 1.6 mEq/liter and to correct the flat T waves. In a short communication, Rosefsky (1972) reported that a premature infant, who had required several exchange transfusions, and who had been given calcium prophylactically, developed arrhythmia with many ectopic ventricular beats during his fourth transfusion. This infant responded to slow intravenous injection of magnesium sulfate (25 to 50 mg/kg) within five minutes, with return of rhythm to normal. When the ectopic beats recurred on subsequent exchange transfusions, intravenous magnesium therapy again rapidly restored the rhythm to normal. ECG changes: high P waves, ST depression, and flat or inverted T waves, are common during exchange transfusions (Robinson and Bathe, 1963). Citrate-lowered ionized magnesium (Bajpai et al., 1967a,b) has been implicated in the high sudden-death rate during this procedure (Editorial, Canad Med Assoc J, 1967).
9.3.2.2. Open-Heart Surgery
The use of ACD blood in the pump oxygenator prime during cardiopulmonary bypass (performed during hypothermic or normothermic anoxic arrest with ventricular fibrillation) has been associated with even more arrhythmias. Scheinman et al. (1969) investigated the pre- and postoperative magnesium status of 17 adult patients undergoing intracardiac operative procedures, after they had observed classic neuromuscular signs of acute magnesium depletion in a patient soon after cardiopulmonary bypass. Levels of three of the patients could not be compared because they had remained in persistent fibrillation, despite standard therapy (including multiple attempts at internal defibrillation) until they were given an intravenous bolus of magnesium sulfate. Of the remaining 14, 12 exhibited drops in serum magnesium from 1.65 ± 0.10 mEq/liter before surgery to 1.07 ± 0.01 mEq/liter after surgery. Despite the hypomagnesemia, no gross neuromuscular abnormalities were noted. This group of investigators then compared those patients with another group of eight, who had magnesium (2 mEq/liter) added to the pump prime (R. Sullivan et al., 1969; Scheinman et al., 1971/1973). Although there was no apparent relationship between postoperative serum magnesium levels and the development of new postoperative arrhythmias, 9 of the 17 patients in the first group developed such arrhythmias, whereas only 2 of the 8 to whose prime magnesium had been added developed arrhythmias. An average of 6.2 shocks at 4 watts/second was necessary for internal defibrillation in the first group versus 1.3 shocks in the second group. The first group of patients had a 30.3% drop in postoperative serum levels versus a 17.4% drop in the second group. Although the fall in serum magnesium was significantly less in the second than in the first group, five of the eight subjects still became hypomagnesemic. The authors suggested that physiologic amounts of magnesium should thus be added to all administered fluids, as well as to the pump prime, and that the magnesium levels should be monitored in all cardiac surgery patients (Scheinman et al., 1971/1973).
Buky (1970) commented that the fibrillation produced during open-heart surgery is a consequence of induced hypothermia, and occurs at an esophageal temperature of about 27° C. (The nature of the pump prime was not noted.) He found that an intravenous bolus of magnesium sulfate (0.1 g/kg intravenously) facilitated postoperative defibrillation, making electrical shocks unnecessary in 18 (66.7%) of the 27 patients so treated, as compared with only eight (19.5%) of the 41 patients not given the magnesium. Spontaneous defibrillation took place when the magnesium level in the serum reached 4.5-5.0 mEq/liter.
Proof that increasing the magnesium/calcium ratio favorably influences the defibrillation threshold was provided by Koning et al. (1971/1973), using dogs on cardiopulmonary bypass. They found that magnesium lowered and calcium increased the pulse amplitude needed for defibrillation. On the basis of their observation, they suggested that it might be useful to administer magnesium to a patient prior to defibrillation.
It has been clearly demonstrated that patients undergoing cardiac surgery lose magnesium, as indicated by increased renal clearance of magnesium despite hypo- magnesemia (Scheinman et al., 1969, 1971/1973) and by serum magnesium levels which are markedly lower during and after the surgical procedure and which remain subnormal for three to seven days postoperatively (Holden et al., 1972; Khan et al. 1973). Even on the day of admission the average serum magnesium levels of the prospective open-heart surgery patients were 10% below normal (1.35 mEq/liter); most with low levels had been on diuretics (Holden et al., 1972). At the end of the operation the average serum magnesium level was 20% below normal (1.1 mEq/liter), and the day after surgery it dropped to 30% below normal (1.04 mEq/liter). It gradually rose thereafter to 5.5 and 1% below normal by the seventh postoperative day and discharge day, respectively. As in the initial study of Scheinman et al.(1969), this group found no definite relationship between the degree of individual lowering of serum magnesium and problem of postoperative arrhythmia. These investigators commented on the role of the citrate in removing ionized magnesium as well as of calcium, on the high flow perfusion in increasing urinary magnesium loss, and on the sharp drop in serum levels despite increased tissue catabolism and hypoxia, which cause shifts of intracellular to extracellular fluids. That patients undergoing open-heart surgery lose myocardial magnesium in the course of the procedure has been demonstrated by Singh et al. (1971/1972). They took myocardial biopsies soon after interruption of coronary flow for about 20 minutes in 16 patients, immediately before reestablishment of coronary flow in 2 and after coronary reflow in 14. They reported that loss of magnesium and potassium from the myocardium was a constant finding, that of magnesium ranging from 2% to 19%.
Khan et al. (1973) gave magnesium supplements to two groups among his 29 open-heart surgery patients: (1) a group of 8 who developed multiple ectopic beats, extrasystoles, and periods of tachycardia postoperatively (100 mg magnesium as the chloride, orally, starting on the third postoperative day), and (2) another group of 8 who were given 100 mg of magnesium (as the chloride) orally pre- and postoperatively and to whose priming fluid 90mg of magnesium (as the sulfate) was added. In contrast to the magnesium levels of nine adults not given magnesium supplements preoperatively, and who had not had magnesium added to the ACD blood prime, who developed definite hypomagnesemia by the end of the procedure, most of those treated prophylactically became only slightly hypomagnesemic after surgery (Fig. 9-3). Also, those given magnesium showed much more rapid return to normal serum magnesium levels. As a result of these findings, Khan et al. (1973) began routine use of 200 to 300 mg of magnesium chloride daily, by mouth, in divided doses, preoperatively, and up to 35mg of magnesium as the sulfate in each 500 ml of dextrose saline solution postoperatively. In addition, they primed the heart-lung machine with 120 mg of magnesium as the sulfate. This regimen keeps the postoperative serum magnesium levels within normal limits. Caution is exercised in the presence of impaired renal function.
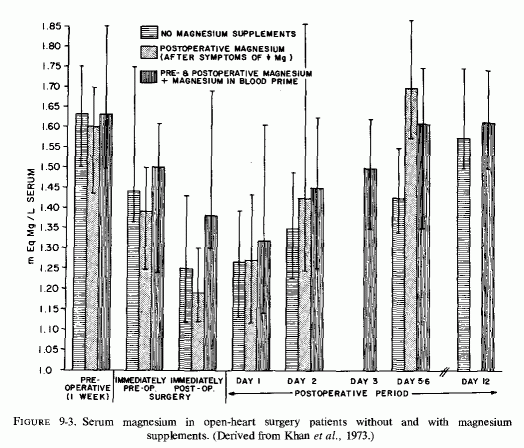{kind=link}
Holden (1978) has recently reported a double-blind study of 70 cardiac surgery patients who were randomly assigned to two postoperative treatment groups: (a) six intravenous doses of 2 ml of a MgSO4 solution containing 0.8 mEq/liter, starting an hour after surgery and thereafter every six hours intramuscularly; and (b) placebo solution of 2 ml normal saline. There was persistent hypomagnesemia for 72 hr postoperatively in the placebo group, and correction of the hypomagnesemia in the group treated with MgSO4 (Fig. 9-4), differences that were significant (p 0.00l) with high confidence limits (90%). Atrial fibrillation had been present in similar numbers in each group preoperatively. Twelve of the preoperative atrial fibrillating patients treated with placebo continued to fibrillate postoperatively, versus one in the magnesium-treated group (Table 9-3). There were more postoperative clinical problems in the group receiving placebo than in those treated with magnesium (Table 9-4). Holden (1978) observed that patients whose plasma magnesium levels were in the normal range were more readily paced than were those whose levels were subnormal. He also commented that, in addition to the patients in his double-blind study, he had encountered 11 (with marginally low mean magnesium level of 1.5 mEq/liter) whose ventricular fibrillation was refractory to conventional treatment for half an hour, and who rapidly responded to intravenous magnesium by return to sinus rhythm or conversion to atrial fibrillation (Table 9-5).
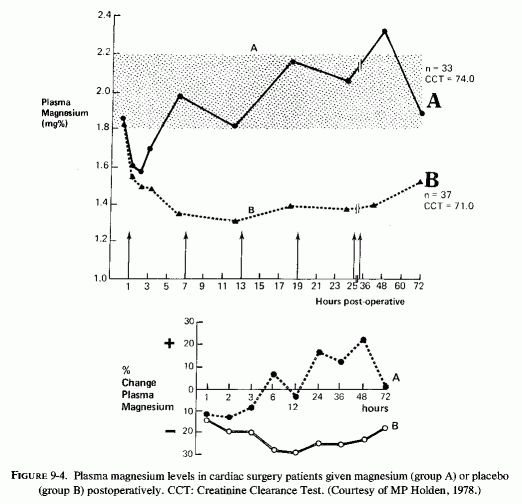{kind=link}
{kind=link}
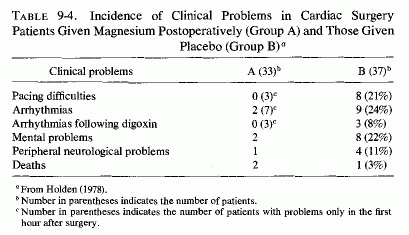{kind=link}
{kind=link}
The development of ischemic contracture of the heart-"stone heart"-during open-heart, cardiopulmonary surgery is rare (Cooley et al., 1972). Of 13 patients (among almost 5000 cardiac procedures in one institution), all had advanced cardiac disease. Twelve had interstitial fibrosis; all had severe myocardial hypertrophy, but only 4 had evidence of recent ischemia. The condition has been totally refractory to reversal. Cooley et al. (1972) suggest that the tetanic contracture might reflect ATP-depletion, or possibly accumulation of calcium, and that catecholamine production (in response to the ischemia) might intensify the situation. Katz and Tada (1972) considered the biochemical mechanism that might be operative in this surgical catastrophe. They point out that ATP can promote contraction (by causing actin and myosin to interact) or relaxation in the presence of increased magnesium concentration. They speculate that the hypertrophied hearts, which might be subject to development of the contracture during surgery, might have been functioning with depleted stores of energy phosphate compounds. Furthermore, such patients are likely to have undergone vigorous diuresis, that leads to metabolic alkalosis (Katz and Tada, 1972), and loss of magnesium (Bajpai et al., 1971/1973; Holden et al., 1972; Lim and Jacob, l972a; Khan et al., 1973; Loeb et al., 1968; Seller et al., 1966; Wacker, 1961), and probably also have received cardiac glycoside therapy, which increases myocardial calcium uptake and loss of myocardial magnesium (Holland, 1964). Since "calcium rigor" has been produced in frogs' hearts, suspended in Ringer's solution with an excess of calcium (Fukuda, 1970), it is possible that addition of magnesium to the preoperative regimen and to the pump prime might function to protect against development of "stone heart."
Prolonged use of magnesium-free parenteral fluids is another cause of acute magnesium depletion that has been associated with arrhythmias, the one that was identified first. Flink et al. (1954) described an ECG, characteristic of hypokalemia, in a patient who had received prolonged parenteral therapy, that was associated with hypomagnesemia and that was corrected by intramuscular magnesium sulfate therapy. It was not until five years later that cardiac irritability, responsive to magnesium therapy, began to be noted in the literature as a risk of surgery, prolonged parenteral therapy, and loss of gastrointestinal fluids, whether from drainage, intractable vomiting, or diarrhea. R. E. Randall et al.(1959) reported several such patients. One had, in addition to neuropsychiatric manifestations of combined hypomagnesemia and hypocalcemia, developed QT prolongation, and depressions of the ST segments and T-wave voltage after infusion with calcium gluconate. Magnesium sulfate was then added to the intravenous fluids, and 18 hours later all of his manifestations of a "terminal" state had cleared. Similar ECG changes were seen in a 38-year-old diabetic man in the Randall et al. (1959) series. This patient had renal wastage of magnesium, and improved somewhat following a 2-week course of parenteral magnesium therapy, only to die of a myocardial infarction a month later. Other patients in this series, whose abnormal ECGs improved with magnesium therapy, had alcoholism or chronic glomerulonephritis. Among the W. O. Smith et al. (1960) series of 18 patients with nonalcoholic neuropsychiatric manifestations of magnesium depletion (10 of whom had sinus tachycardia and sometimes frequent premature ventricular systoles) were 3 who had had prolonged infusions, 1 who had long-term severe diarrhea, and 4 with acute pancreatitis. Hanna et al. (1960) reported 3 patients with ECG signs of magnesium depletion: low voltage of all complexes, which increased following treatment with magnesium chloride. One of their patients had had malabsorption and had been given very high doses of vitamin D, had another for renal osteomalacia. The third was hypomagnesemic immediately following parathyroidectomy. Baron (1969) reported a patient who developed magnesium-responsive tachycardia after surgery and prolonged parenteral fluids.
The ECG changes of a patient who had undergone extensive surgery and received intravenous fluids and had gastrointestinal suction following a complicated postoperative course were reported in detail by Kellaway and Ewen (1962). When her serum magnesium level was 1.3 mEq/liter, she exhibited flattened T-wave and ST depression that were apparent particularly in the chest leads, but also in the standard leads (Fig. 9-5). Magnesium sulfate (20%) was added to the iv. fluid and given at the rate of 8 mEq/hour. Within 24 hours, her ECG had returned to normal (Fig. 9-6). In addition to her moderate hypomagnesemia, this patient had slightly higher than normal plasma potassium (5.7 mEq/liter), but because her ECG was more like the tracings seen in severe hyperkalemia, the authors considered the hypomagnesemia contributory. They suggested that serial electrocardiography might be a helpful adjunct in controlling electrolyte replacement therapy. Five years after the published case report, the patient was seen by Dr. Kellaway, who reported her in good health and with a normal ECG (personal communication).
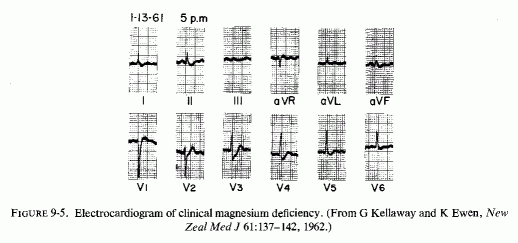{kind=link}
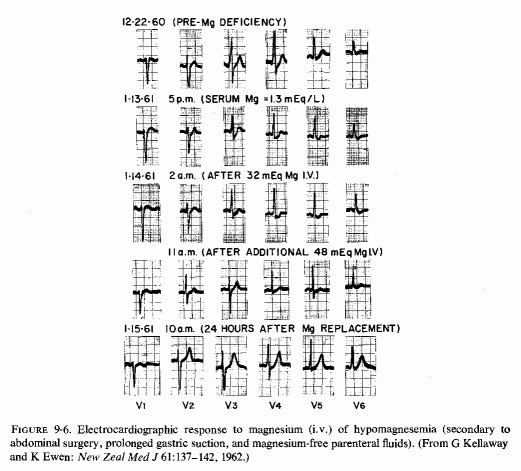{kind=link}
Thoren (1963), who presented a detailed biochemical and surgical report of 15 patients with magnesium deficiency secondary to losses of gastrointestinal fluids, reported the ECG of only one. That patient (with prolonged biliary drainage) first had an ECG described as typical of hypokalemia, despite ample potassium supplements, and then developed postoperative tachycardia, intraventricular conduction block, and unspecific ST-T changes. W. O. Smith (1963) reported 7 patients with postoperative, neuropsychiatric signs of magnesium deficiency, all but one of whom had been on constant gastric drainage and all of whom had been on magnesium-free intravenous fluids for 1 to 90 days with little or no oral intake. Of this group, whose serum magnesium values ranged from 0.50 to 1.29 mEq/liter, tachycardia, acute hypertension, or both were seen in four. Treatment with magnesium intravenously or intramuscularly, as recommended by Flink (1956), produced marked improvement in all cases within 4 to 24 hours. In a brief case report of a patient with ulcerative colitis, who had a preoperative ECG suggestive of hypokalemia, and who ha been intensively treated with blood transfusions, ACTH, oxytetracycline as well "vigorous" potassium therapy, Matko (1966) mentioned ST-T abnormality and typical neuropsychiatric signs of acute magnesium depletion after a stormy postoperation course, necessitating gastric suction and continuous intravenous feeding. The body magnesium stores of this patient must have been severely lowered, in view of her illness, that was associated with long-term diarrhea, the administration of ACTH and blood (probably citrated), and even the tetracycline, which chelates and inactivates magnesium (Shils, 1962). Yet her serum magnesium level at the height of her signs of depletion was only moderately low (1.2 mEq/liter). She responded promptly, with subsidence of all of the signs of magnesium depletion, after having had four grams of magnesium sulfate (32.5 mEq magnesium) given intravenously in 250 ml 5% dextrose in water over 2-hour period.
It is not possible to assess the frequency of magnesium-deficiency-induced ECG abnormalities developing in patients who have undergone major surgery and/or had drainage and prolonged parenteral fluids. Henzel et al. (1967), who reviewed the risks and consequences of magnesium deficiency in surgical patients, referred to low voltage ECG, tachycardia, and premature ventricular contractions as manifestations that might develop, and cited the special risk of any potential surgical candidate whose abnormal nutritional status might lead to magnesium deficiency. What is needed is a systematic survey of the magnesium status of such patients for ECG abnormalities, and for their response to magnesium therapy, with electrocardiographic monitoring, as recommended by Kellaway and Ewen (1962).
The literature reviewed has uncovered few patients with malabsorption syndromes, in which electrocardiographic changes have been attributed to magnesium depletion, to which this group of patients is particularly susceptible. However, hypokalemic and hypocalcemia ECGs have been attributed to gastrointestinal disorders that lead to losses of potassium and calcium (Lepeschkin, 1959). It is likely that magnesium depletion participates as well, contributing both to potassium loss (Review; Seelig, 1972) and to hypocalcemia (Review: Massry, 1977). Furthermore, severe hypomagnesemia causes ECG tracings resembling a combination of characteristics seen with potassium and calcium depletion.
The patient reported by Gerst et al. (1964), who developed severe hypomagnesemia (0.37 mEq/liter), with confusion, tremors, convulsions, and a sinus tachycardia of 140/minute, had had several segmental bowel resections and long-term loss of intestinal fluids from his stoma. This patient showed clearing of sensorium and tremors, with subsidence of tachycardia within six hours of receiving 4 grams of MgSO4 (40 ml 10% solution of 500 ml of dextrose and water). Bajpai et al. (1971/1973) have reported the ECG changes of two patients with severe steatorrhea and malabsorption, who had almost as low serum magnesium levels: 0.62 and 0.82 mEq/ liter. They both had tachycardia, low PQRS voltages, and flat to inverted T waves. These patients were given oral magnesium chloride supplements (124 mEq magnesium/day, added to the standard hospital diet) for 21 days. This treatment corrected the hypomagnesemia and resulted in improved PQRS voltage and T waves (Fig. 9- 7). Lim and Jacob (1972d) reported low voltage and flat T waves in the ECG of three of seven patients with chronic diarrhea, two of whom had hypomagnesemia (1.4 mEq/liter and 1.1 mEq/liter), but all of whom had subnormal skeletal muscle magnesium levels.
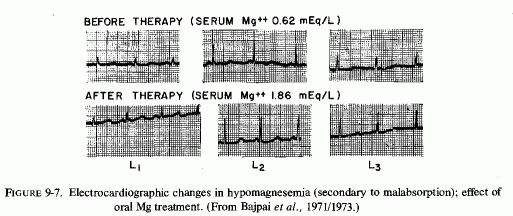{kind=link}
Previously undetected malabsorption and steatorrhea was identified in a 72- year-old woman, who was admitted to the hospital with a history of sudden onset of palpitations and syncope the day before and two hours before admission (Chadda et al., 1973b). She had premature ventricular systoles and minor ST abnormalities. She then developed supraventricular tachycardia, and was found to have hypokalemia (3.0 mEq/liter). Intravenous potassium chloride raised the blood levels to normal, but did not correct the arrhythmia. She was then found to have severe hypomagnesemia (0.35 mEq/liter). During the next paroxysm of supraventricular tachycardia with aberrant conduction, an intravenous injection of 2 ml 25% magnesium sulfate caused prompt return to sinus rhythm. She was then given a constant infusion of 1 g magnesium sulfate (8 mEq Mg) per hour for 12 hours, at which time her serum magnesium level became normal. After diagnosis of malabsorption, she was given oral and intramuscular magnesium supplements for a year and had no recurrence of syncope or cardiac arrhythmia.
The syndrome, protein caloric malnutrition (PCM), is an example of gastroenteritis in babies and young children in which magnesium-responsive ECG changes have developed during the "recovery syndrome." Caddell (1965) reported ECG changes, such as have been reported in severe experimental magnesium deficiency, in PCM children fed high-protein milk with added potassium and sodium chloride. She compared the ECGs of 103 affected children on the standard regimen with and without magnesium supplements. On admission, most had sinus tachycardia (130 to 160/mm); some had bradycardia. On admission the rate was usually very labile. P waves were small, absent, or notched; the PR interval was short, and there were dwarfed QRS complexes, ST abnormalities and flat or inverted T waves in limb leads I and II and in left precordial leads V5 and V6. Intermittent gallop rhythm occurred in four, one of whom developed ventricular ectopic beats the day before death. The children given magnesium supplements usually showed some lengthening of the previously fixed short PR interval, and greater increases in the T-waves amplitude than were seen during the recovery phase of the nonsupplemented children. The improvement of the other complexes was not specifically attributed to the magnesium. In later studies, Caddell (1967, 1969a,b) reaffirmed and extended her observations in the ECG abnormalities of PCM, and their response to magnesium therapy. She found the PCM children with flat or inverted T waves over the precordium on admission generally grew worse on the standard therapy. After five days treatment, there was further inversion of the T waves, development of labile heart rate, and rarely ventricular ectopic beats. These children were usually hypotensive, and tolerated blood transfusions and digitalis poorly. The transfusions often led to congestive heart failure; the digitalis given in the usual therapeutic dose usually led to severe arrhythmias and gastrointestinal disturbances. When magnesium (0.5 mEq/kg) was given intramuscularly, clinical improvement was noted within six hours. The precordial impulse improved, the cardiac sounds became louder and of better quality, and a stable normal sinus rhythm developed (Caddell, 1969b).
Caddell made an interesting observation that seems worth exploring. She commented that survivors of PCM often have persistent PR interval and T-wave abnormalities, that endomyocardial fibrosis is found in the same geographic regions as PCM, and that the morphology of the cardiac lesions resemble those that Selye (1958f) reported to be protected against by magnesium and potassium. She speculated, thus, that the ECG abnormalities of PCM might reflect mineral imbalance, and that persistent deficiencies of magnesium and potassium might be contributory to the development of endomyocardial fibrosis (Caddell, 1965).
Hypocalcemic ECG tracings were obtained from a baby with isolated magnesium malabsorption that did not respond to calcium but improved once high magnesium requirements were met (Nordio et al., 1971).
9.3.4. Arrhythmias of Starvation
Sustained loss of magnesium from lean tissue and bone has been reported among volunteers (nonobese) who have undergone short- to long-term (45 days) periods of starvation. Keys et al.(1950) has reported slight prolongation of the QT interval in volunteers who underwent prolonged starvation, and suggested that this might indicate myocardial damage.
Obese patients who fast to lose weight are at even greater risk, both because they lose substantial amounts of magnesium and because they mobilize body fat, which increases risk of magnesium depletion (Consolazio et al., 1967; Jones et al., 1969: Drenick et al., 1969; Drenick and Brickman, 1971/1973). A dramatic case that had ECG signs suggestive of magnesium deficiency was a twenty-year-old woman who died on the seventh day of refeeding after 30 weeks of starvation, and was found to have cardiomyopathy at autopsy (Garnett et al., 1969). She had normal-serum electrolytes except for one episode of hypokalemia, at which time the ECG showed SF depression and slight QT prolongation. She was given potassium supplements, despite which she had further loss of exchangeable potassium (from 3360 Eq to 1400 mEq), indicating loss of lean mass tissue. After her cardiac arrest, which responded to cardiac massage, she had an obviously prolonged QT interval and T-wave inversion in leads I, aVL, and aVR. She was given a slow intravenous potassium infusion, which was stopped when the report of plasma potassium of 3.7 mEq/liter was received. Her serum levels of magnesium and calcium were 2.25 and 8.4. She was then given 1 g of calcium chloride in 5% dextrose in an hour, at which time she developed multifocal ventricular extrasystoles, and lignocaine was substituted for the calcium. She again had an episode of ventricular fibrillation seven hours later, her QT interval remained lengthened despite having received 30 mEq of potassium overnight, and she died of ventricular fibrillation refractory to electrical defibrillation. The authors had considered the possibility of magnesium deficiency, but were reassured by the normal serum magnesium level. Fasting patients have maintained normal or even elevated serum magnesium levels despite sustained loss of magnesium stores (Sunderman, 1947; Sunderman and Rose, 1948). Whether this patient would have survived had she been given magnesium chloride, rather than potassium and calcium, cannot be averred. In view of its cardioprotective effect, however, it is suggested that this approach, possibly with potassium chloride, might be worth trying. Another obese patient, one who had been dieting on a liquid-protein diet supplemented with vitamins, calcium, and potassium, and who developed syncope and arrhythmia, had a low serum magnesium level (1.5 mEq/liter) that was considered marginally normal; she was given a constant infusion of magnesium (dosage not specified). Her serum magnesium level never rose to over 1.67 mEq/liter, and she died of ventricular fibrillation and cardiomyopathy (Michiel et al. 1978).
9.3.5. Arrhythmias of Alcoholism
Tachycardia and hypokalemia-like ECG changes were observed by Flink and his collaborators (1954, 1957) and by Smith and Hammarsten (1959) in patients with alcohol withdrawal symptoms and signs, which responded to magnesium therapy. It is noteworthy that arrhythmias and ECG changes reported in heart disease of chronic alcoholism are often found in association with hypomagnesemia (Flink et al., 1954, 1957; Randall et al., 1959; Fankushen et al., 1964; Milner and Johnson, 1965; Loeb et al., 1968; Hartel et al., 1969; Ricketts et al. 1969; Bajpai et al.1971/1973; Iseri et al., 1975; Iseri and Bures, 1976/1979). Alexander (1968), who had noted the similarity of the ultramicroscopic changes seen in alcoholic cardiomyopathy and those seen in magnesium-deficient animals (Alexander, 1966a,b) tabulated the ECG findings in alcoholic heart disease from a series of 127 admissions for 66 patients (Table 9-6). Brigden and Robinson (1964), who had earlier considered it likely that the magnesium loss caused by alcoholism might be contributory to the cardiomyopathy of alcoholism, also referred to the wide range of ECG abnormalities found. Among his 50 patients, who had consumed large quantities of alcohol, tachycardia was present whether or not there was sinus rhythm. Ectopic beats were common and often multifocal. Half had atrial fibrillation at some time; it was more common in the older patients. Abnormalities of the T waves occurred in most of the ECGs. Conduction defects were present in 19 of the patients. The authors noted that the ECG findings are dependent on the site and extent of myocardial damage: A small area of damage strategically placed causes a more significant conduction defect than a similar lesion deep in the muscle mass. U-wave abnormalities, such as are seen in hypokalemia, were reported also in the ECG of a young man with alcoholic heart disease, who had hypomagnesemia (0.75 mEq/liter) but normal potassium serum levels (Ricketts et al., 1969). He also had alternating upright and inverted T waves, and the more common sinus tachycardia. These abnormalities were intermittent and cleared during hospitalization even though he was not given magnesium therapy and received digitalis and diuretics. T-wave alternans was reported in a man with long-lasting heavy drinking, who had hypomagnesemia when admitted (1.14 mEq/liter). The T-wave abnormality disappeared after three days of small i.v. doses of magnesium (Luomamaki et al. 1975)
{kind=link}
The study by Härtel et al. (1969) in Finland showed less frequency of abnormal ECG findings (Table 9-7) than did the American and English studies reported by Alexander (1968) and Brigden and Robinson (1964). They commented that the frequency of ECG abnormalities was about the same in the alcoholics as it was in other groups of Finnish men of the same age. They commented that the pattern of drinking in Finland differed from the chronic alcoholism of the patients in Alexander's (1968) and Brigden's and Robinson's (1964) studies, in that 80% of their patients were intermittent drinkers, eating amply between "binges." A point requiring clarification is whether the ECG abnormalities found in nonalcoholic Finns (in rural areas) might be related to the high incidence of sudden death from ischemic heart disease in north and eastern Finland. The study by Härtel et al.(1969) was done in Helsinki, in the southeast of Finland. In that study, however, 42% of their patients had hypomagnesemia, but they did not correlate the ECG abnormalities with the low serum levels of magnesium, except for its occurrence in 3 of their 7 patients who had prolonged QT intervals. They confirmed the observation of T. James and Bear (1967) that the sinus tachycardia seemed to be mediated by catecholamines, since it was depressed in 17 of 21 patients by β-adrenergic blockade. Although acetaldehyde perfusion of the sinus node of dogs caused sinus tachycardia (James and Bear, 1967), and abnormal alcohol metabolism to acetaldehyde was therefore implicated, it is possible that magnesium-deficiency-induced catecholamine release might be contributory, since magnesium deficiency from many causes has also caused tachycardia and ECG changes, such as are seen in alcoholism.
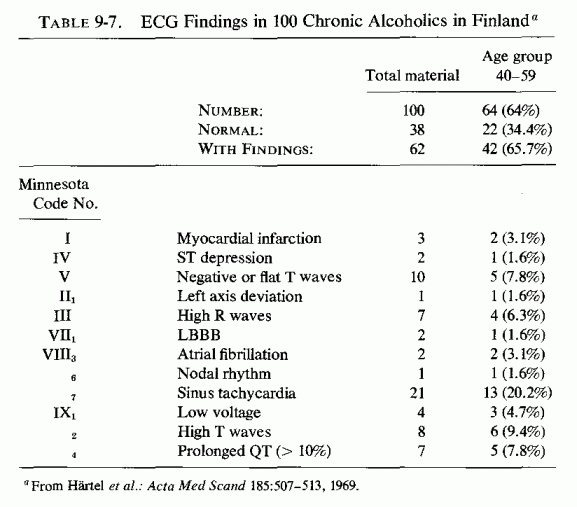{kind=link}
Only rarely has the electrocardiographic response of alcoholic arrhythmia to magnesium been recorded. The tracing by Benchimol and Schlesinger (1953) is included in Fig. 9-8 (line C), with the proved Mg-depletion ECG of Kellaway and Ewen (1962) given for comparison in line A. Randall et al. (1959) mentioned that his patients with ECG abnormalities (QT prolongation, depressed ST segment and T wave), several of whom were chronic alcoholics with and without cirrhosis, showed improvement of all signs, including the abnormal ECOs when they were treated with magnesium sulfate. One of the hypomagnesemic patients of Fankushen et al. (1964) had sinus tachycardia and a prolonged QT interval that persisted after all of the previously abnormal serum electrolytes had returned to normal, except her hypomagnesemia. She was then given magnesium supplements, with resultant elevation of her serum magnesium to normal, whereupon she again began to drink heavily. Loeb et al. (1968) reported an alcoholic young woman with paroxysmal tachycardia and serum magnesium levels of 0.5-0.7 mEq/liter. She had an ECG pattern characterized by QT prolongation preceding appearance of bigeminy, multifocal ventricular complexes, and ventricular fibrillation terminating spontaneously in sinus rhythm. Despite her severe hypomagnesemia, which was associated with episodes of syncope and tonic convulsions, she was not given magnesium, but treated traditionally for her cardiac problem with procainamide, dephenylhydantoin, potassium, calcium chloride, and 50% dextrose-all of which were ineffective. The arrhythmia finally responded to artificial cardiac pacing.
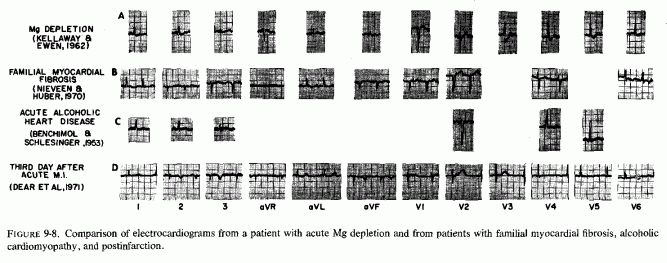{kind=link}
Among the hypomagnesemic patients of Bajpai et al., (1971/1973) with characteristic ECG abnormalities that responded to rapid magnesium injections (80 mEq as the sulfate in 15 ml 25% glucose) were four with decompensated hepatic cirrhosis (etiology not designated), whose hypomagnesemic ECG changes developed after furosemide treatment. Within 15 min of the magnesium injection, at a time that the serum magnesium had risen to 1.85-2.05 mEq/liter, the ECGs showed rapid increases in PQRS voltages and lesser increases in T waves. With slow oral magnesium replenishment (as in their patients with malabsorption), the T-wave voltage improved as well (Fig. 9-9).
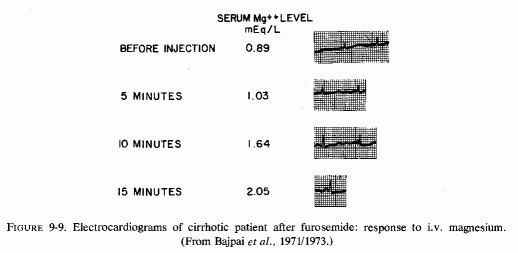{kind=link}
Until Iseri et al. (1975, 1978) reported electrocardiographic improvement on magnesium therapy of two alcoholic patients with arrhythmias refractory to standard treatment, the hypomagnesemia of alcohol withdrawal has been commonly disregarded, as improving on ethanol discontinuation and resumption of normal diet. Iseri et al. (1975) concerned themselves with the probable cellular magnesium deficiency of these patients and included magnesium in their treatment regimen. Their first patient had sinus tachycardia and deeply inverted T waves on admission. She then developed ventricular fibrillation, was countershocked and given lidocaine and procainamide, which was effective for about 12 hours. Recurrent episodes of ventricular fibrillation were repeatedly treated similarly. When she was given 10 ml of 20% magnesium sulfate over a 1-min period, the fibrillation was abolished. (Serum magnesium, taken before the magnesium bolus was given, was 1.39 mEq/ liter.) Sinus rhythm was maintained with infusion of lidocaine (2 mg/min) and magnesium (20 mg/min) and oral quinidine at 200 ml every 4 hours. After she had received 15 g of magnesium sulfate by infusion over an 8-hr period, she again developed ventricular tachycardia. It did not respond to lidocaine but did to another 10 ml intravenous injection of 20% magnesium sulfate. She was given 5 g more of the magnesium sulfate, had the lidocaine stopped, but was continued on oral quinidine and given potassium chloride for rapidly developing hypokalemia. Their second patient had congestive heart failure, accelerated junctional rhythm, and flat T waves. After digitalis and furosemide, he developed atrial tachycardia and multiple ventricular beats and runs of ventricular tachycardia, resistant to lidocaine. He was treated with magnesium, as had been the first patient, and his ventricular arrhythmia was immediately abolished. Both patients had been treated with digitalis: the first had levels that were well below toxic; the second had digitoxicity, as well as a history of chronic alcoholism. The authors considered the refractory arrhythmia of both patients to be secondary to magnesium depletion and that the second case was complicated by digitoxicity. (They reviewed the literature on the role of magnesium loss, caused by digitalis and diuretics in cardiac patients.) They recommended magnesium therapy for the treatment of cardiac arrhythmias, whether alcohol- or digitalis-induced, or spontaneous. The general regimen recommended is 10-15 ml of 20% magnesium sulfate intravenously over 1 min, followed by a slow 4- to 6-hour infusion of 500 ml 2% magnesium sulfate in 5% dextrose in water, the infusion to be repeated if arrhythmia recurs.
Flink (1969) formulated a magnesium-treatment program for the hypomagnesemia of alcoholism, which should be applicable to the cardiomyopathy and ECG abnormalities of alcoholism, as well as to the more commonly reported neuropsychiatric manifestations. He suggests continuous intravenous infusions for 48-60 hours, providing 50 to no more than 100 mEq of magnesium every 12 hours, or 16 mEq of magnesium (2 g of 50% MgSO4 solution intramuscularly) every two to six hours for about five days. In 1969, Flink's group suggested that it is at least as appropriate to replace magnesium by the parenteral route in chronic alcoholism as it is to replace potassium or other electrolyte deficits. Flink (1976/1980) expressed surprise that, of the nutrients known to be deficient in alcoholics, magnesium alone is rarely considered in replacement therapy. As recently as September, 1977, Fisher and Avrams described the response of an alcoholic with tachyarrhythmia to low dosage MgSO4 (1 ml every 6 hours) plus procainamide, but commented that although hypomagnesia of alcoholism is well-documented, its replacement remains controversial. This evoked letters to the editor from Flink (1978) and Moore (1978), who reiterated the importance of adequate magnesium repletion.
Disease of cardiac-conducting tissue is more frequent in diabetic patients than in other disease categories (Rubler et al., 1975). Among 45 patients with idiopathic complete or partial heart block, 25% were known diabetics, and 34% more had abnormal glucose levels. McMullen (1977) reported an adolescent diabetic girl who developed sudden asystolic arrest while her diabetes was improving on conventional treatment. After reinstituting cardiac activity by classic procedures, she was found to have severe hypomagnesemia (0.6 mEq/liter). She was immediately repleted with 120 mEq of magnesium i.v. over the next 6 hours; there was gradual return of normal cardiac rhythm without further antiarrhythmic drugs.
Toxemia of pregnancy is a condition in which magnesium deficiency has been implicated and in which ECG changes not unlike those seen in severe magnesium deficiency have been recorded (Fig. 9-10). The illustration depicts ECGs from patients who developed heart failure toward the end of pregnancy or in the early postpartum period. The first report of ECG changes found were in 1937 (Gouley et al.); inversion of T waves was described. Hull and Hakfesbring (1937) and Hull and Hidden (1938) commented the most common abnormality seen in postpartal heart failure is low or inverted T waves, and that gallop rhythm is common. Thomson et al. (1938) reported that most toxemic patients had abnormal T waves, and that even 7.7% of normal pregnant women had abnormal T waves in a chest lead, that reverted to normal some time after delivery. Dexter and Weiss (1941) commented that the heart was usually normal in mild toxemia but found postpartum ECG abnormalities in 2 of 12 patients on days 6 and 9, respectively. One of them had exhibited hypertension and developed heart failure 7 days before term, but had a normal ECG; 9 days postpartum the tracing showed inverted T waves in leads I and IVF. Dieckman (1942) concurred that patients with mild toxemia rarely show cardiac damage, but found that those with severe preeclampsia and eclampsia usually had tachycardia and sometimes developed heart failure. Freundlich (1946) reported tachycardia and ventricular extrasystoles in a woman with a negative cardiac history until the birth of her second child. In their study of ECGs of 12 women with toxemias of pregnancy, Wallace et al. (1946) reported less severe ECG changes in 4, who did not develop heart failure, than in 2 who went into postpartum cardiac decompensation. One toxemic woman had T-wave inversion; a comparable tracing was obtained from 1 of 5 women who had normal pregnancies. They suggested that the focal myocardial necrosis that is sometimes seen in women with toxemic pregnancies is a probable cause of the T-wave abnormalities, and might be a factor in postpartum cardiac failure. Szekely and Snaith (1947) found ECG abnormalities in 7 of 19 unselected (most severe) cases of toxemia of pregnancy. They exhibited transient alterations of the T waves, usually in both standard and chest leads, similar to those seen in anterior myocardial infarction (and similar to magnesium deficiency ECG). Sinus tachycardia was frequent, and 2 had extrasystoles. Three of the patients had left ventricular failure, and the authors considered the electrocardiographic changes in at least 5 indicative of myocardial damage. Although the cardiac changes seemed to be temporary in most, their duration varied considerably. and sometimes worsened in the postpartum period. Melvin (1947) noted QRS complexes of low amplitude and low voltage or inverted T1, T2, and T 4 and sinus tachycardia in four patients with postpartum heart failure, with a definitely prolonged QT interval in one. Decherd and Henmann (1944) reported supraventricular tachycardia in a woman with cardiac failure 2 months after premature termination of her second complicated pregnancy, and commented on the rapidity with which her tachycardia stopped (temporarily) when her circulation time was tested with a magnesium sulfate injection. Serial ECGs were obtained in 10 of 15 patients with myocardial failure developing in the last trimester of pregnancy and the puerperium (Meadows, 1957). In each instance, the admission ECG showed T wave inversion in multiple limb and precordial leads. None showed significant Qwaves or conduction defects. In 5 patients, the ECGs became normal within 1-7½ months, and some improvement was seen 9-19 months later in 4. Preeclampsia had been diagnosed in only 3 of this series of 15 patients with peripartal cardiomyopathy. Seftel and Susser (1961) found normal ECGs in only 3 of 23 patients in Africa with peripartum cardiac failure.
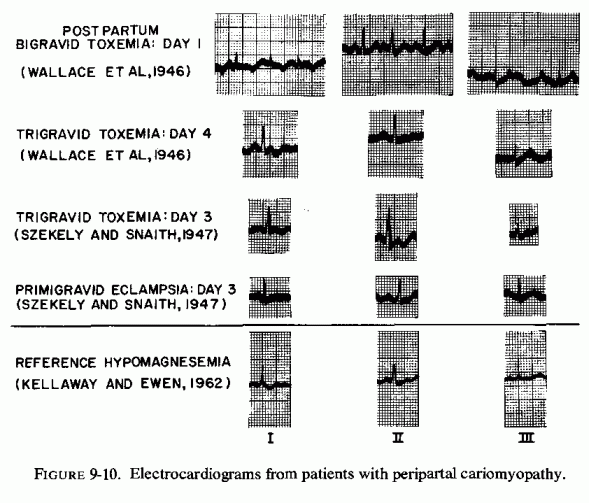{kind=link}
J. B. Johnson et al. (1966) found low QRS voltage and absent or inverted T waves in the limb leads, and discordant T waves in leads V1-V6 in a 14-year-old mother of twins who had cardiomyopathy diagnosed by biopsy four months after delivery, following cardiac decompensation that developed during her third trimester. She was very sensitive to digitalis toxicity and died seven months after delivery.
Walsh et al. (1965) reported transitory rhythm disturbances: bigeminy, trigeminy, and multiple unifocal and multifocal premature ventricular contractions in a series of 15 patients in Jamaica, most of whom were malnourished young multipara.
Left bundle branch block, frequent extrasystoles, and P-R prolongation were seen in some or all of 7 (out of 10) patients with cardiomyopathy of pregnancy and the puerperium reported by Stuart (1968). This investigator commented that the changes are indicative of focal myocardial damage, and are in keeping with the frequent occurrence of angina in patients with peripartal cardiomyopathy with often persistent ECG abnormalities (Meadows, 1957; Gilchrist, 1963), and with the high prevalence of angina and electrocardiographic evidence of focal myocardial lesions in patients from the same population group (Jamacia) with idiopathic cardiomegaly (Stuart and Hayes, 1963; Fodor et al., 1964). Sakakikibara et al. (1970) reported right bundle branch block, abnormal Q waves, and flattened or inverted T waves in a woman with postpartum cardiomyopathy, confirmed by electron microscopy of a biopsy specimen.
Ledingham et al. (1968), after reporting a young woman who suddenly developed a cerebrovascular accident while under observation for minor antepartum bleeding, and who died despite heroic measures (including caesarean section in a hyperbaric chamber), commented on the desirability of ECG screening of pregnant patients. Their patient had an enlarged heart, triple rhythm, sinus tachycardia, and typical ECG abnormalities of cardiomyopathy of pregnancy, which was confirmed at autopsy. Despite her antemortem findings suggestive of cardiac disease, she had been considered in good health before hospital admission, and the initial ECG (only lead LI) showed only sinus tachycardia. The investigators doubted that their patient's stroke was caused by an embolus; they considered it more likely to have resulted from acute cerebral perfusion failure that might have been caused by severe hypotension associated with transient arrhythmia. They urge screening all pregnant women with 12-lead electrocardiography, to detect all unexplained abnormalities, and to arrange for immediate hospital admission if early signs of decompensation occur.
This is an excellent suggestion, that should be modified by inclusion of screening for occult magnesium deficiency by testing for percentage-retention of a parenteral load of magnesium. Whether infantile and peripartal cardiomyopathies would be reduced in incidence by treatment of pregnant women whose magnesium retention indicates deficiency should be studied. Until definitive results are obtained, this is a benign means of therapy that should be tried once ECG abnormalities are detected. Reference should be made here to the improved maternal response and fetal salvage of eclamptic women treated with magnesium salts, as compared with those treated with diuretics, sedatives, or antihypertensives (Zuspan and Ward, 1965).
Congenital electrical disturbances of the heart would be expected to be manifest very early in life. Possibly arrhythmias might contribute to the sudden-infant-death syndrome (SIDS). T. James (1976, 1968) and Ferris (1972, 1973) have proposed that a contributory abnormality might be damage to the small coronary arteries that cause myocardial damage involving the conducting system of the heart. In a provocative brief report of examination of myocardial and conduction tissue of infants who had died of SIDS and of 22 control infants (who had died of traumatic, infective, or other identified cause), W. Anderson et al. (1970) found degenerative changes in areas including portions of the A-V node and bundle of His in all of the infants. An infant born with A-V block, who soon developed congestive heart failure and died at two months of age, had degenerative changes and calcification in the central body of the A-V bundle, fibrosis in the left bundle branch, and subendocardial calcification adjacent to the right bundle branch (R. A. Miller et al., 1972). Kariv et al. (1964, 1971) observed that the familial form of cardiomyopathy, which begins very early in life, is characterized by arrhythmia, and that this suggests very early development of histopathologic changes. Arteriosclerosis of large and small coronaries, and focal myocardial necrosis and fibrosis, have been found in infants who died suddenly and in others who had been ill with clinically manifest heart disease, many of whose first cardiac manifestations developed at about two to four months of age, the age of peak incidence of SIDS. It seems possible that a factor that might cause fatal arrhythmia and cardiac arrest in some infants might cause silent cardiac damage permitting longer survival in some, and "benign" silent arrhythmias in others, depending on the area affected and complicating factors (e.g., infection, congestion, and their treatment).
ECG evidence of myocardial ischemia, but no reported coronary lesions, has been reported among infants with "primary" or idiopathic myocardial disease (Freundlich et al., 1964). Paroxysmal atrial tachycardia and arrhythmias, including conduction blocks, multiple premature ventricular beats, and intraventricular conduction disturbances, with and without familial or isolated cardiomyopathy, has been reported in infancy (Freundlich et al., 1964; Kariv et al., 1964; Lev et al.. 1967; Simcha and Bonham-Carter, 1971; Haese et al., 1972; Bove and Schwartz.1973). The pattern of arrhythmias of the familial form of cardiomyopathy, beginning very early in life, has suggested early development of histopathologic changes (Kariv et al., 1964).
That conduction defects can develop prenatally is demonstrated by bradycardia and A-V dissociation that had been found in utero in two infants who died with focal myocardial necrosis within three days of birth (Oppenheimer and Esterly, 1967). Paroxysmal atrial tachycardia (PAT) developed at two months of age in a previously healthy infant (Bove and Schwartz, 1973). Digoxin converted the condition to normal sinus rhythm but preexcitation persisted despite continued digitalization. She developed normally for the next half year until three days before her death. Her PAT recurred and she was given intramuscular digitalis and morphine, to which the PAT did not respond. Intravenous phenylephrine was started and promptly stopped when multiple premature ventricular contractions developed. Ventricular fibrillation ensued, which responded to a single direct current shock, after which she developed bradycardia with ventricular bigeminy followed by brief periods of junctional tachycardia with premature ventricular contractions. She then returned to supraventricular tachycardia. On transfer to the Medical Center, she repeatedly required direct current .cardioversion. She remained stable for 14 hours with a cardiac rate of 140/min after a pacing wire was placed. When ventricular fibrillation recurred, she developed profound hypotension, for which she was given intravenous infusions of isoproterenol and epinephrine. She died 48 hours after admission and was found to have cardiomegaly and lipid cardiomyopathy. The baby reported by Lev et al. (1967) developed a conduction defect at 12 1/2 months, after having been alert and healthy for his first year of life. He died after a month of hospitalization, during which he exhibited intermittent 2:1 block with Adams-Stokes episodes. At autopsy, he had severe intimal proliferation encroaching on the lumen and degenerated and partially calcified elastica of the main coronary arteries and some of the branches. There were no atheromata. Numerous large and small recent, organizing, and old infarcts were present throughout the septum and free wall of the left ventricle. Atrioventricular dissociation and ventricular arrhythmias suddenly developed in a 16-month-old girl, who had been in excellent health (Haese et al. 1972). During the ensuing 18 days, she had repeated episodes in association with vomiting, pallor, and cyanosis before dying with multifocal premature ventricular contractions. No light microscopic lesions of the conduction system were found, but there were numerous random foci of myocardial degenerative changes. Endocardial fibroelastosis has also caused comparable arrhythmias: heart block, atrial fibrillation, nodal tachycardia, frequent ventricular premature contractions, flattened or inverted T wave (Moller et al., 1964).
ECG tracings from a 39-year-old man, who died 2 years later of familial myocardial fibrosis, disclosed auricular flutter and occasional ventricular ectopic beats with flat and negative T waves in several leads (Nieveen and Huber, 1970).
9.3.9. "Idiopathic" and Postinfarct ECG Abnormalities That May Be Related to Magnesium Deficiency or Loss
9.3.9.1. "Benign" Arrhythmias
Many cardiac electrical disturbances occur without detectable heart disease (Kastor, 1973). Their cause is unknown. Among the abnormalities reported in hearts diagnosed as "healthy" are paroxysmal atrial fibrillation (Peter et al., 1968), supraventricular tachycardia (Cass, 1967), bundle-branch blocks (Beach et al., 1969; R. F. Smith et al., 1970), "benign" premature ventricular beats and parasystoles (Myburgh and Lewis, 1971), and even some cases of paroxysmal ventricular tachycardia (Lesch et al., 1967). Kastor (1973), who presented this complex of "benign" electrical disturbances, commented that people with such abnormal ECGs cannot be distinguished from normal subjects by any specific pathological abnormality. He suggested that reentrant arrhythmias with preexcitation [ Wolff-Parkinson-White (W-P-W) syndrome] might represent a form of congenital heart disease, as might cases of sinus node disorders (Spellburg, 1971). He points out that fibrosis of the peripheral bundle blocks might be responsible, and that since it occurs in the absence of evidence of coronary artery, myocardial, or infectious disease it, like the other cited "benign" arrhythmias, can be categorized only by the functional disorder, or the presence of fibrosis of unknown cause. The tachyarrhythmias of the W-P-W syndrome are predominantly paroxysmal supraventricular tachycardia (in 70-80% of the cases) and atrial flutter fibrillation (Tonkin et al.., 1976).
The similarity of the "benign" arrhythmias of unknown cause (Kastor, 1973) and that seen in the patient with familial myocardial fibrosis with those of experimental and clinical magnesium deficiency is provocative. Comparison of the tracings obtained from Kellaway's and Ewen's (1962) patient during her acute magnesium depletion and that of Nieveen and Huber (1970) show remarkable similarities. Very-thick-walled arterioles were seen in a muscle biopsy taken from the patient with familial myocardial fibrosis (Nieveen and Huber, 1970). Possibly he also had comparable arteriolar thickening, with a small lumen/wall ratio in the myocardium, as has been reported in magnesium deficiency and in infants and children from ethnic groups with high suspectibiity to early IHD. T. James (1967) has noted the similarity of the lesions of those with pathology of the small coronary arteries to those seen in magnesium deficiency and has considered the possibility that small coronary artery disease, as it causes loss of more and more small foci of the myocardium, may play a role in the pathogenesis of arrhythmias, conduction abnormalities, and sudden death in young victims. He pointed out that if the damaged myocardial foci involve conductive tissue, arrhythmias can ensue. It is well to recall, here, that the interventricular septum has a high magnesium concentration, most of which is readily exchangeable, and which can thus readily be mobilized from the heart, particularly under conditions of stress.
Intramyocardial occlusive coronary lesions and chronic inflammatory microlesions of the myocardium have been found with high frequency in adults who died suddenly and unexpectedly of ischemic heart disease (IHD), as compared with chronic IHD and controls: p < 0.001 (Haerem, 1975). These myocardial lesions were considered likely to render the myocardium especially vulnerable to disturbances of coronary blood flow. Haerem (1975) found that fibrous lesions in the conduction nodes were comparable in the 46 sudden death patients and the 21 patients who died with chronic coronary disease. However, nonfibrous lesions of the atrioventricular node or the bundle of His predominated among the sudden death victims (p < 0.05).
There is substantial attention being paid to the occurrence of fatal IHD without postmortem evidence of occlusive arterial disease (Review: Bajusz, 1965b; Baroldi, 1969, 1970/1972). Also there is increasing evidence that clinically suspect IHD need not be corroborated by demonstrable or sustained occlusion or narrowed major coronary arteries (Dear et al., 1971; Neill et al., 1972; Oliva et al., 1973; R. Henderson et al., 1973; Khan and Haywood, 1974; Brest et al., 1974; Maseri et al., 1975; Haywood et al., 1976), in some instances in teenaged boys (Kimbris et al., 1972; Sidd et al., 1970). Intermittent coronary spasms have been implicated, ever since Prinzmetal et al. (1960) described the variant form of angina that occurs at rest. However, most patients with this disorder have severely narrowed major coronary arteries (MacAlpin, 1973; Arnett and Roberts, 1976). Similarly, necropsy studies have generally confirmed that acute myocardial infarction is associated with old coronary atherosclerotic plaques. Arnett and Roberts (1976) have considered factors that might explain normal (major) coronary arteries in patients who have had infarctions. They commented that patients with myocardial scars despite normal coronaries fall into several groups; (1) those that have left ventricular outflow obstruction (whose lesions are usually subendocardial or in the left papillary muscles), (2) whose intramyocardial arteries only were affected, or (3) who had had an embolus that subsequently lysed or recanalized. The first two categories fit well into the manifestations seen in experimental magnesium deficiency and in many cases of infantile cardiovascular disease that resemble the lesions of magnesium deficiency.
Bajusz (1965b) criticized the classical correlation of myocardial infarction or angina pectoris with anatomical (i.e., major coronary) arterial lesions as mechanistic and one-sided. He proposed that chronic coronary lesions might act as conditioning factors that predispose to myocardial necrosis by metabolic derangements that lead to electrolyte shifts in the myocardium. Such shifts, which are similar in diverse cardiomyopathies, all of which are characterized by myocardial magnesium loss as a consistent early finding (Lehr et al., 1976/1980), can explain the similarity to magnesium deficiency ECGs of electrocardiographic tracings in diverse clinical conditions (including those of chronic ischemia and those seen several days after acute infarction).
It has been proposed that long-term magnesium deficiency can contribute to the atherosclerotic process. The higher incidence of sudden death from IHD in soft-water areas (low magnesium) than in hard-water areas that has been correlated with lower myocardial magnesium levels in accident victims in soft- versus hard-water areas (T. Anderson et al., 1973, 1975, 1978) indicates that low magnesium levels might predispose to sudden arrhythmias. Among survivors of an acute ischemic event, further loss of myocardial magnesium (enhanced by hypoxia and stress hormones) can intensify the problem. The ECG taken at the time of the acute ischemia does not resemble that seen in acute or chronic magnesium deficiency. However, in a recording on the third day of hospitalization for an acute myocardial infarction, the first 48 hours of which had been complicated by frequent premature ventricular contractions (PVCs), episodes of sinus arrest, and A-V dissociation, the ECG resembled strongly that of the patients with magnesium depletion [Figure 9-8D (Dear et al., l97l)]. The correlation of the PVCs and conduction disturbances in this patient, with the development of an ECG that resembled that of severe magnesium deficiency, might be relevant to the observation that patients with postinfarction PVCs have poorer prospects for survival than do those without premature beats. A three-year survey of over 2000 survivors of acute myocardial infarction (Coronary Drug Research Project, 1973) showed that those who had any PVCs had twice the mortality rate of those without that arrhythmia. Since such arrhythmias are seen in magnesium deficiency, and since magnesium has been shown to be cardioprotective, use of pharmacologic doses of magnesium immediately after the acute event, followed by long-term prophylactic supplemental doses, should be tried and systematically investigated.
In this volume we discuss the high incidence of not only osteodystrophy but of metastatic calcification in hemodialyzed uremic patients and the suggestion that use of physiologic concentrations of magnesium in the dialysate (rather than the more commonly used low-magnesium water) might protect against such ectopic calcification. Calcific cardiomyopathy and fibrosis, involving the atrioventricular node, has resulted in heart block in such patients (R. Henderson et al., 1971; Terman et al., 1971; Arora et al., 1975). The magnesium concentration of the dialysate was not given, and in only one report (of six patients with severe myocardial calcification) were serum predialysis (but not postdialysis levels) reported (Terman et al., 1971). The predialysis serum levels in that study were only slightly elevated, but in view of the authors' published concern that high magnesium bone levels in renal disease patients might contribute to osteodystrophy (Alfrey and Miller, 1973), it is probable that low magnesium-water had been used for dialysis. Whether use of magnesium concentration of 1.5 mEq/liter in the dialysate would protect against metastatic calcification as has been proposed by Posen and Kaye (1967), Kleeman et al. (1970), and Danesh et al. (1970), and whether such cardiac damage and dysrhythmias might thereby be averted in patients receiving long-term dialysis requires study.
=======================================
No comments:
Post a Comment